Huilin's Notebook
- MSCA PhD student, Protein research group, Prof.Ole, Synnansk Universitet, Denmark
- Research assistant, Neurodegenerative Lab, Westlake University, China
- Master degree, Computer Science: Data Science, Leiden University, the Netherlands
- Bechalor degree, Information management and Information system, Huazhong Agricultural University, China
Binder Design
target.pdbpreprocessing- binders design via RFdiffusion and ProteinMPNN-FastRelax
- backbones filtering
- pae_interaction score
OVO
install
- https://ovo.dichlab.org/docs/user_guide/installation.html
conda create --name myovo python=3.13conda activate myovopython --version(Python 3.13 recommended, at least 3.10)java -versionJava (OpenJDK 21-24 recommended, at least 17)
module load jdk/18.0.1.1
pip install ovoovo init home
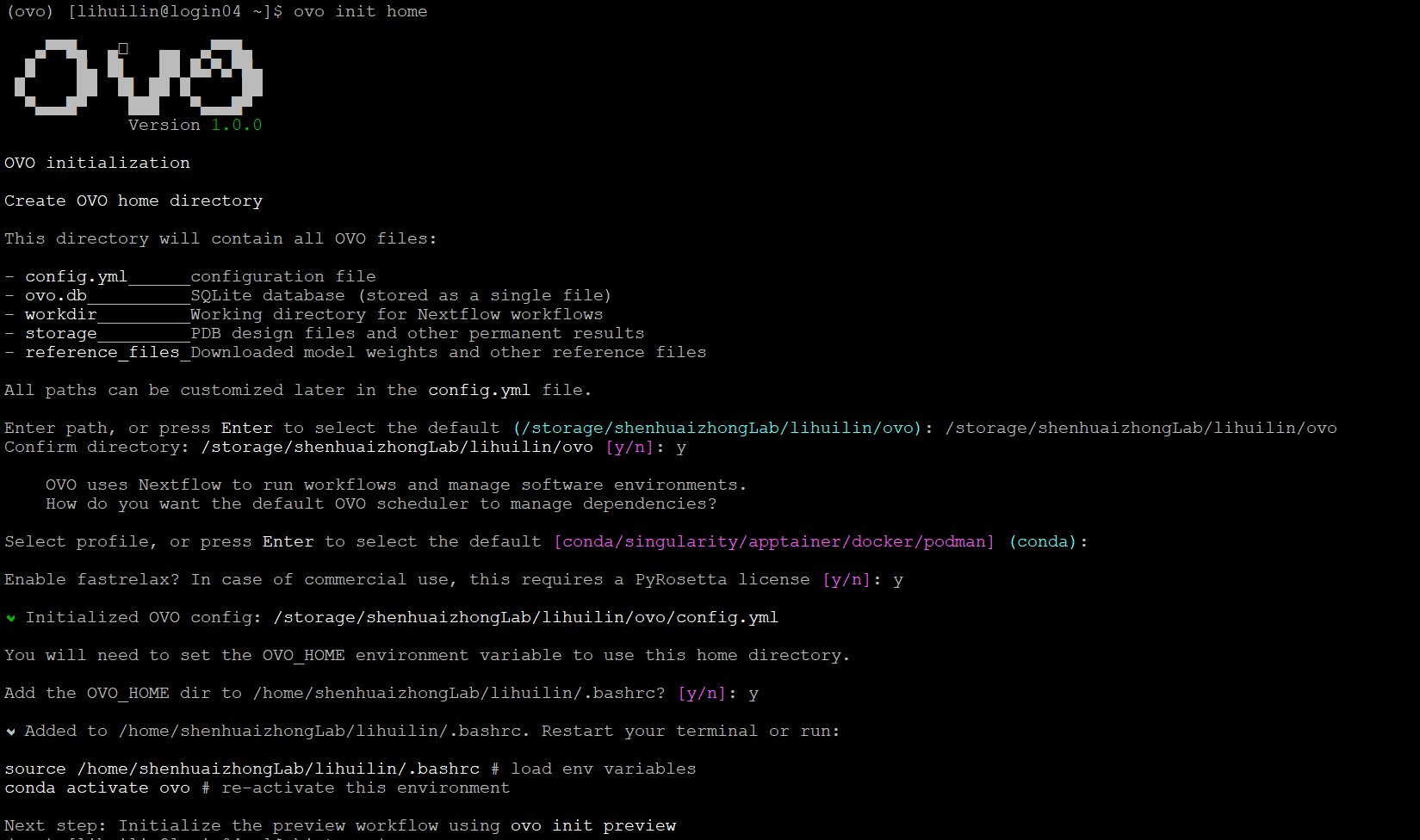
source /home/shenhuaizhongLab/lihuilin/.bashrc# load env variablesconda deactivateconda activate myovo# re-activate this environmentconda install -c bioconda nextflow
(myovo) [lihuilin@login02 ~]$ nextflow -version
N E X T F L O W
version 25.10.2 build 10555
created 28-11-2025 19:24 UTC (29-11-2025 03:24 CDT)
cite doi:10.1038/nbt.3820
http://nextflow.io
ovo init preview
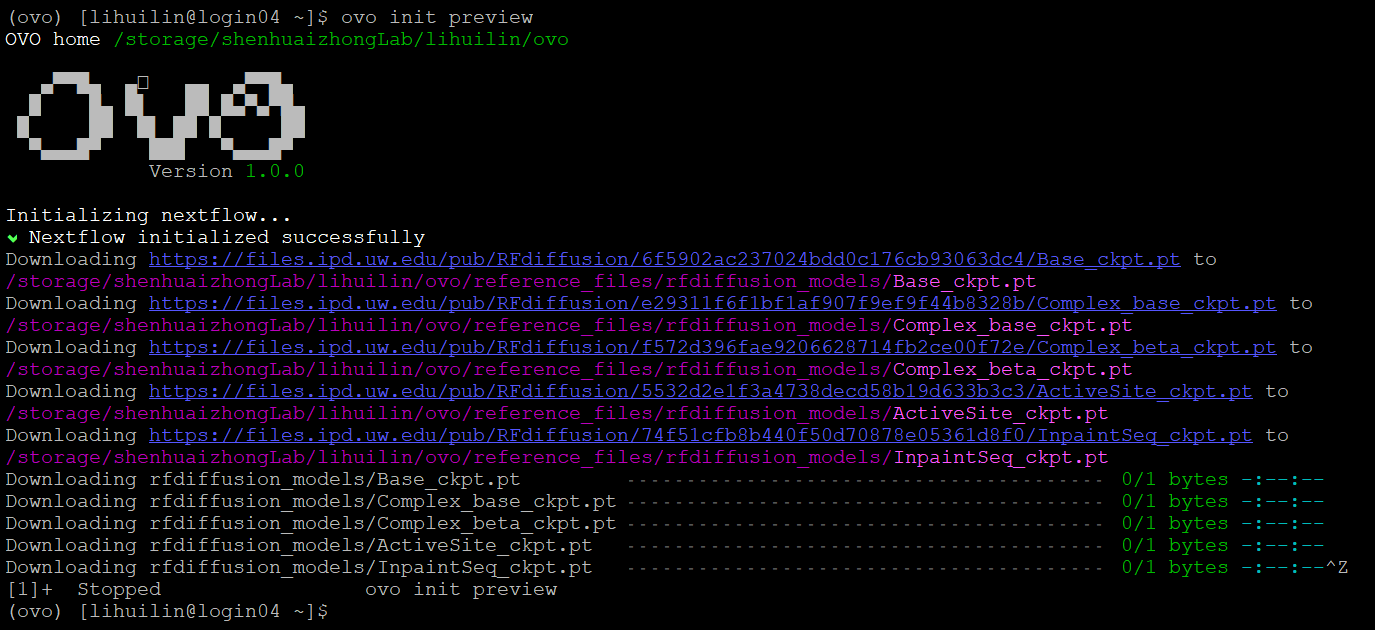
Download RFdiffusion model checkpoint files:
In practice, due to the network constraints on HPC, it will be much faster to download these files locally, and then transfer them to the correct folder on HPC. (e.g. /storage/shenhuaizhongLab/lihuilin/ovo/reference_files/rfdiffusion_models)
- (optional)
Process requirement exceeds available CPUs -- req: 4; avail: 1
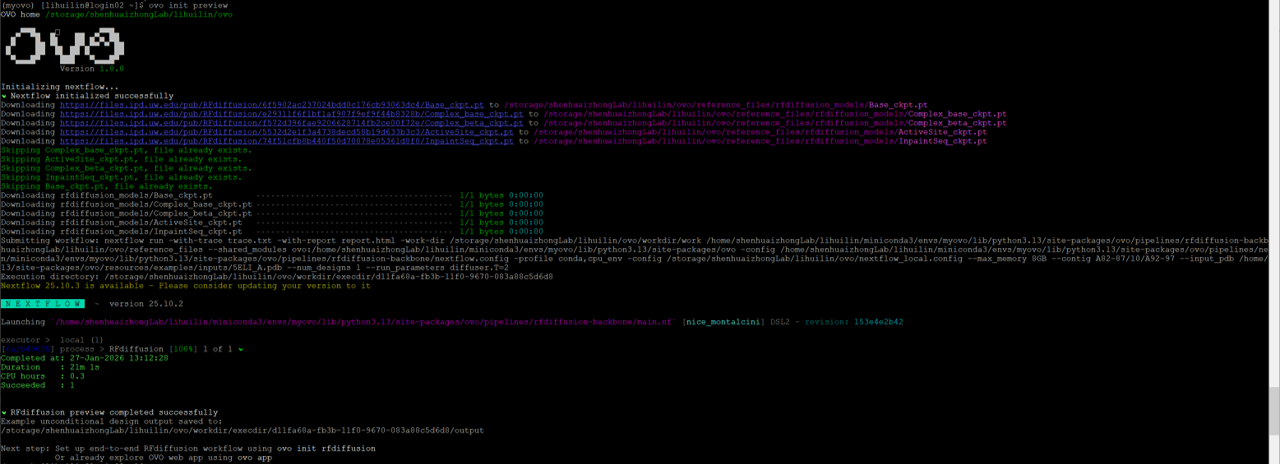
ovo init preview requires internet accession and CPUs\(\ge\)4 simultaneously. In my practice, the login node can access internet, but only have 1 CPU. In this case, we can modify /home/shenhuaizhongLab/lihuilin/miniconda3/envs/myovo/lib/python3.13/site-packages/ovo/pipelines/rfdiffusion-backbone/main.nf file by changing cpus 4 to cpus 1 (line 11) and memory "16 GB" to memory "8 GB" (line 12).
ovo init preview
ovo app
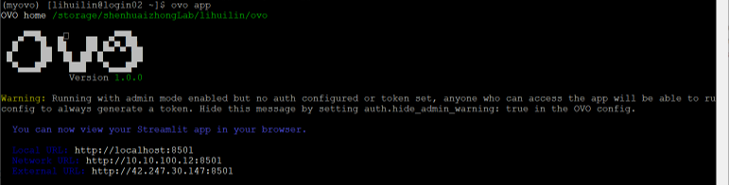
ssh -L 8501:localhost:8501 lihuilin@172.16.78.132 -p 10002
target.pdb preprocessing
Assuming we have a target protein target.pdb, and we want to create a binder to target.pdb. The first step is preprocessing target.pdb. Generally spearking, there are two steps we need to think of.
Truncating target.pdb
Truncating target protein helps a lot in reducing the stress of computing. For example, we could remove AAs with very low pLDDT or AAs who are far away from the place we want binders to bind with.
def residue_remove(pdbfile, remove_ids, outputName):
"""
Remove residues based on positions.
Assumption: pdbfile only has A chain
"""
residue_to_remove = []
pdb_io = PDB.PDBIO()
pdb_parser = PDB.PDBParser()
structure = pdb_parser.get_structure(" ", pdbfile)
model = structure[0]
chain = model["A"] # assuming pdbfile only has A chain
for residue in chain:
id = residue.id
if id[1] in remove_ids:
residue_to_remove.append(residue.id)
for residue in residue_to_remove:
chain.detach_child(residue)
pdb_io.set_structure(structure)
pdb_io.save(outputName)
return
# truncating ids
truncating_ids = [1,2,3]
# remove some AAs
residue_remove(pdbfile="target.pdb", remove_ids=truncating_ids, outputName="truncation.pdb")
This step can also be achieved in PyMol.
How many AAs we can remove from our target protein?
At the very beginning, I was thinking removing all useless AAs. For example, I was creating a binder to a transmembrane protein, and I only left the part of protein which is located at the outside of the whole protein. Unfornutately, this part of protein I aim to create binders to bind with is too "thin" to lead design binders correctly.
If the "target" protein you feed into the RFdiffusion model is too small, ppi.hotspot_res argument becomes ineffective. Let's imagine the "target" protein we feed into the RFdiffusion model is a paper, and ppi.hotspot_res is located somewhere in this paper. Since this "target" protein (the paper) is too thin, RFdiffusion model feels confused about which side (above or below the paper ) the ppi.hotspot_res points to.
My suggestion would be to try some different trunctations and pick one that can approcimately balance the computation stress and correct leading of bidner design.
Reset AAs' residues numbers in truncation.pdb
Normally, after removing some AAs, the residues number in truncation.pdb becomes discontinuous, and we want to concatenate them again and update their residue number with continuous numbers.
def update_resnum(pdbfile):
"""
🌟 CHANGE RESIDUE NUMBER
Assumption: pdbfile only has A chain
"""
pdb_io = PDB.PDBIO()
pdb_parser = PDB.PDBParser()
structure = pdb_parser.get_structure(" ", pdbfile)
model = structure[0]
chain = model["A"]
print(len([i for i in chain.get_residues()]))
new_resnums = list(range(1, 1+len([i for i in chain.get_residues()])))
for i, residue in enumerate(chain.get_residues()):
res_id = list(residue.id)
res_id[1] = new_resnums[i]
residue.id = tuple(res_id)
pdb_io.set_structure(structure)
pdb_io.save(pdbfile.split('.pdb')[0] + '_update_resnum.pdb')
return
Design binders via RFdiffusion and ProteinMPNN-FastRelax
In my use case, firstly, I tried to de novo design binders. However, I found that designed binders are limited to proteins with only one helix. Then, I changed to design binders based on scaffolds. The design process and scripts I used a lot are as follows.
Three needed designing environments
- RFdiffusion for desigining protein backbones, under
SE3nv environment. - ProteinMPNN-FastRelax for desiging protein sequences, under
dl_binder_design environment. - AlphaFold2 for evaluating designed proteins, under
af2_binder_design environment.
We will design binders under these three environments: SE3nv environment, dl_binder_design environment, af2_binder_design environment.
Install these three environments Make sure you have high performance computating envrionment with CUDA, and you can access GPU and internet. Their installations can automatically install dependencied packages that fit the GPU environment which you have to use for the heavy computation.
Quickly go through the whole process
Let us say that we want to design binders for target.pdb, and our current directories tree is
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── target
│ │ └── target.pdb
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
1️⃣ get secondary structure and block adjacency information script get_adj_secstruct.slm
We will provide these two .pt files (target_adj.pt and target_ss.pt) to the scaffold-based binder design model.
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -o %j.out
source ~/miniconda3/etc/profile.d/conda.sh
conda activate SE3nv
cd /(root)/mydesigns
python3 ../RFdiffusion/helper_scripts/make_secstruc_adj.py --input_pdb ./target/target.pdb --out_dir ./target_adj_secstruct
Then, our directories tree is updated as:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
2️⃣ backbones design script bb.slm
I use array-job to parallelly execute different ppi.hotspot_res arguments simultaneously. For example, I have 10 ppi.hotspot_res arguments to test, so my paraID.txt is like:
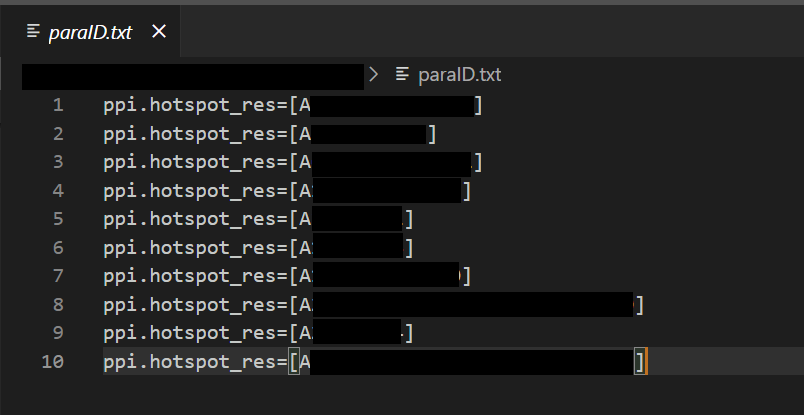
and I setup 10 jobs (#SBATCH -a 1-10) for each ppi.hotspot_res. Each ppi.hotspot_res will design inference.num_designs=10000 binder backbones.
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -J array-job
#SBATCH -a 1-10
#SBATCH -o backbone_10000.%A.%a.log
id_list="./paraID.txt"
id=`head -n $SLURM_ARRAY_TASK_ID $id_list | tail -n 1`
source ~/miniconda3/etc/profile.d/conda.sh
conda activate SE3nv
cd /(root)/mydesigns
python3 ../RFdiffusion/scripts/run_inference.py \
scaffoldguided.target_path=./target/target.pdb \
inference.output_prefix=backbones_OUT/ \
scaffoldguided.scaffoldguided=True \
$id scaffoldguided.target_pdb=True \
scaffoldguided.target_ss=./slightly_truncation_adj_secstruct/slightly_truncation_ss.pt \
scaffoldguided.target_adj=./slightly_truncation_adj_secstruct/slightly_truncation_adj.pt \
scaffoldguided.scaffold_dir=../RFdiffusion/examples/ppi_scaffolds/ \
scaffoldguided.mask_loops=False \
inference.num_designs=10000 \
denoiser.noise_scale_ca=0 \
denoiser.noise_scale_frame=0
Untar 1000 scaffold templates
Don't forget to untar the provided 1000 scaffold templates (RFdiffusion/examples/ppi_scaffolds_subset.tar.gz). In ppi_scaffolds_subset.tar.gz, there is ppi_scaffolds folder and we will use it in our backbone designing script.
Now, our directories tree becomes:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
Add ppi_scaffolds argument in backbone name
In RFdiffusion/scripts/run_inference.py, I modify some codes a little bit, so that, the backbone pdb file name will contain the `ppi_scaffolds` argument.
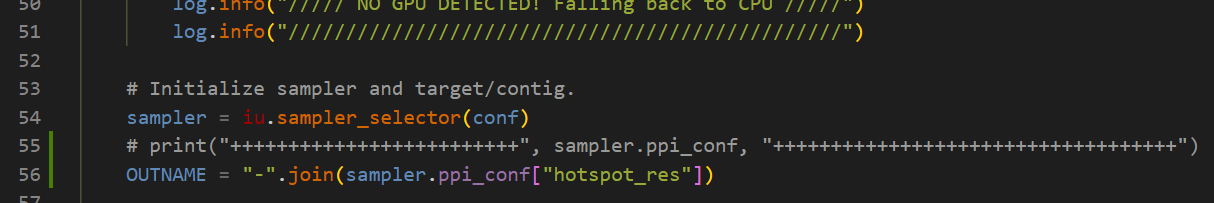

inference.num_designs=10000 binder backbones for ppi.hotspot_res=[A28-A25-A29-A26-A63] argument is A28-A25-A29-A26-A63_0.pdb.
3️⃣ sequence design and binder assessment script mpnn_af.slm
Perform a filtering step
At this moment, we could perform a filtering step after we get all potential binder backbones. For example, I only want backbones (orange cross) in one side (green circle). We could remove other useless backbones, which can help a lot in reducing the computation stress.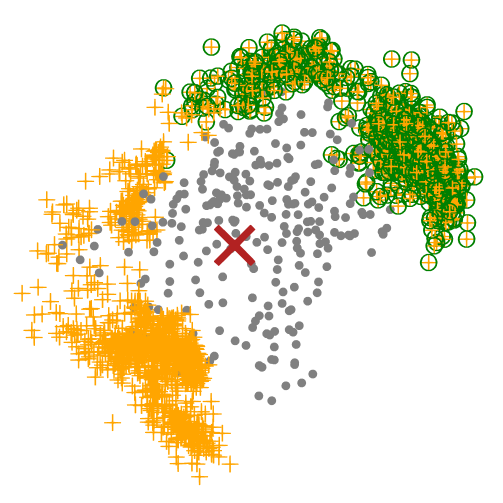
Let's say that we finally select 1000 favourite backbones, and we go to design binders based on these 1000 backbones. Normally, I will split favourite backbones into multiple folders, so I could parallelly design binders in each folder simultaneously. For example, in order to speed up the binder design process, I split the 1000 favourite backbones into 5 folders. After that, I could parallely execute #SBATCH -a 1-5 jobs simultaneously.
# sele_list contains names of 1000 selected backbones,
# such as A28-A25-A29-A26-A63_0.pdb
folder_size = 100 # how many pdb files you want at most to be grouped in one folder
chunks = [sele_list[x:x+folder_size] for x in range(0, len(sele_list), folder_size)]
# len(chunks)=5
for i in range(len(chunks)):
chunk = chunks[i]
newpath = "../mydesigns/select_1000_mpnn_af/folder" + str(i)
if not os.path.exists(newpath):
os.makedirs(newpath)
for j in chunk:
src = "../mydesigns/backbones_OUT/" + j
dst = "../mydesigns/select_1000_mpnn_af/folder" + str(i) + "/" + j
shutil.copyfile(src, dst)
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -J array-job
#SBATCH -a 1-5
#SBATCH -o mpnn_af_1000.%A.%a.log
id_list="./folderID.txt"
id=`head -n $SLURM_ARRAY_TASK_ID $id_list | tail -n 1`
cd /(root)/mydesigns/target_1000_mpnn_af
cd $id
source ~/miniconda3/etc/profile.d/conda.sh
conda activate proteinmpnn_binder_design
silentfrompdbs *.pdb > silent.silent
python ../../../BinderDesign/dl_binder_design/mpnn_fr/dl_interface_design.py -silent silent.silent -outsilent seq.silent
conda deactivate
conda activate af2_binder_design
python ../../../BinderDesign/dl_binder_design/af2_initial_guess/predict.py -silent seq.silent -outsilent af2.silent -scorefilename score.sc
Now our directories tree is like:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
│ ├── select_1000_mpnn_af
│ │ ├── folder0
│ │ │ └── A28-A25-A29-A26-A63_0.pdb
│ │ │ └── ...
│ │ │ └── af2.silent
│ │ │ └── check.point
│ │ │ └── score.sc
│ │ │ └── seq.silent
│ │ │ └── seq.silent.idx
│ │ │ └── silent.silent
│ │ │ └── silent.silent.idx
│ │ ├── folder1
│ │ ├── ...
│ │ ├── folderID.txt
│ │ ├── mpnn_af.slm
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
4️⃣ Get designed binders
In each mydesigns/select_1000_mpnn_af/folder, af2.silent has our designed binders. And, we will extract their .pdb files from the .silent file by (for example)
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
cd /(root)/mydesigns/select_1000_mpnn_af/folder0
source ~/miniconda3/etc/profile.d/conda.sh
conda activate proteinmpnn_binder_design
silentextract af2.silent
Our final directories tree is like:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
│ ├── select_1000_mpnn_af
│ │ ├── folder0
│ │ │ └── A28-A25-A29-A26-A63_0.pdb
│ │ │ └── A28-A25-A29-A26-A63_0_dldesign_0_cycle1_af2pred.pdb
│ │ │ └── ...
│ │ │ └── af2.silent
│ │ │ └── check.point
│ │ │ └── score.sc
│ │ │ └── seq.silent
│ │ │ └── seq.silent.idx
│ │ │ └── silent.silent
│ │ │ └── silent.silent.idx
│ │ ├── folder1
│ │ ├── ...
│ │ ├── folderID.txt
│ │ ├── mpnn_af.slm
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
Each file with _cycle1_af2pred.pdb in name is our expected designed binders.
pae_interaction score
As RFdiffusion states, we expect a binder whose pae_interaction score is lower than 10. However, we should also have a look at other scores, such as pLDDT, rmsd.
check_n = 6
folder_nam = ["folder"+str(i) for i in range(check_n)]
score_paths = ["../MyBinderWork/ThreeThoudand_mpnn_af/"+nam+"/score.sc" for nam in folder_nam]
ALL_DF = []
for p in score_paths:
df = pd.read_csv(p, delim_whitespace=True)
ALL_DF.append(df)
SCORE_df = pd.concat(ALL_DF)[["binder_aligned_rmsd", "pae_binder", "pae_interaction", "plddt_binder", "description"]]
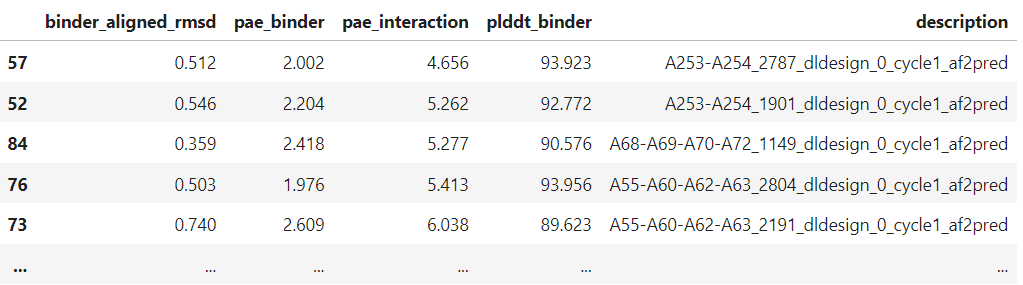
backbones filtering
In design binders via RFdiffusion and ProteinMPNN-FastRelax, at the beginning of sequence design and binder assessment script mpnn_af.slm, we could perform a fitering step to remove useless backnones.
In my opinion, this is a very heloful step, because it greatly increase the speed and efficiency of desigining reasonable binders. For example, in my target.pdb, I only expect designed binders could bind with the outside part of the target.pdb (see fig.1).
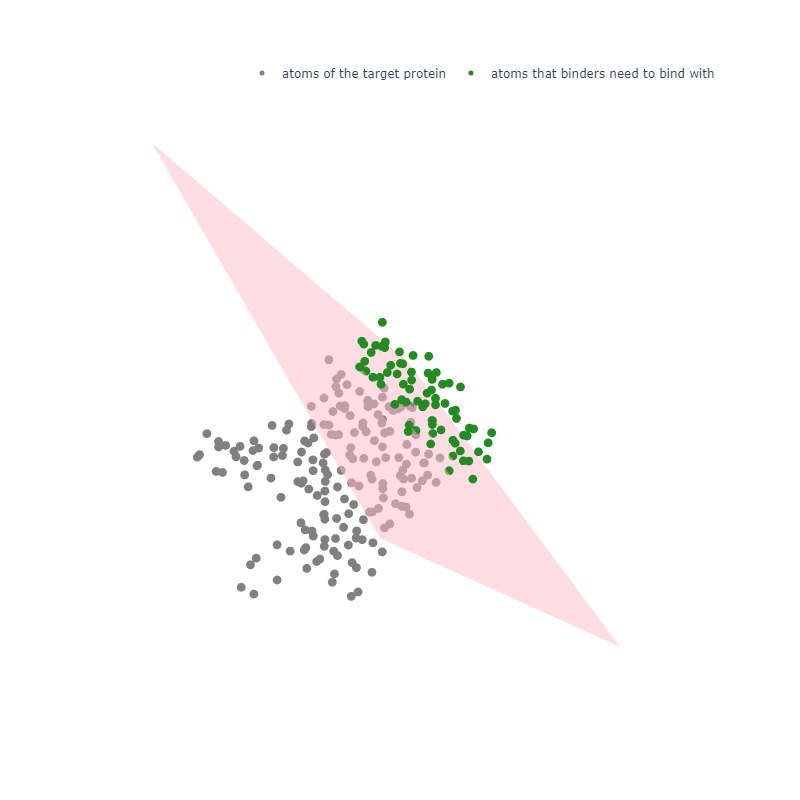

results
- RFdiffusion is better at designing binders for target proteins that are already existing or have been verified by experiments. If the target protein is predcited by AI models, and has not been verified by experiments, the probability of designing a potential binder for this target will be reduced.
- The number of designed binders should be huge, in order to pick potential candidates as many as possible.
CompBio
- PARSE nodes.dmp
- PARSE json from foldseek search server
Environment Setup
Important setup:
torch, torch_geometric and their dependencies: torch_scatter and torch_cluster
-
conda create -n plienv python=3.9.18 -
nvcc -V
nvcc: NVIDIA (R) Cuda compiler driver
Copyright (c) 2005-2024 NVIDIA Corporation
Built on Thu_Sep_12_02:18:05_PDT_2024
Cuda compilation tools, release 12.6, V12.6.77
Build cuda_12.6.r12.6/compiler.34841621_0
python
Python 3.9.18 (main, Sep 11 2023, 13:41:44)
[GCC 11.2.0] :: Anaconda, Inc. on linux
Type "help", "copyright", "credits" or "license" for more information.
>>>
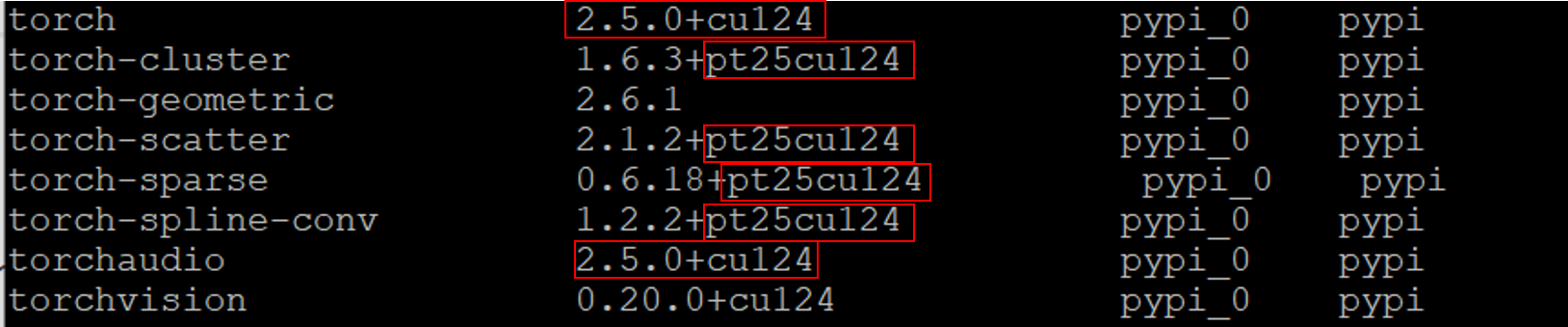
- https://pytorch.org/get-started/previous-versions/
pip install torch==2.5.0 torchvision==0.20.0 torchaudio==2.5.0 --index-url https://download.pytorch.org/whl/cu124
- https://pytorch-geometric.readthedocs.io/en/latest/install/installation.html
pip install torch_geometric
pip install pyg_lib torch_scatter torch_sparse torch_cluster torch_spline_conv -f https://data.pyg.org/whl/torch-2.5.0+cu124.html
IMPORTANT:
-
Both PyG and PyTorch must be installed via
pip -
PyTorch and CUDA version must be same.
-
PyTorch installation must be through Wheel not Conda.
-
PyTorch 2.5.1 doesn't work with PyG 2.5.0.
pip install e3nnpip install fair-esmpip install rdkit-pypipip install biopythonpip install ProDypip install matplotlibpip install pandaspip install PyYAML
Chapter 4 The Taxonomy Project
https://www.ncbi.nlm.nih.gov/books/NBK21100/
Each entry in the database is a “taxon”, also referred to as a “node” in the database. The “root node” (taxid1) is at the top of the hierarchy.
The path from the root node to any other particular taxon in the database is called its “lineage”; the collection of all of the nodes beneath any particular taxon is called its “subtree”.
https://ftp.ncbi.nih.gov/pub/taxonomy/
- taxdump.tar.gz
- taxdump.tar.gz.md5
which contain MD5 sums for the corresponding archive files. These files might be used to check correctness of the download of corresponding archive file.
gunzip -c taxdump.tar.gz | tar xf -
taxonkit
conda install -c bioconda taxonkit
taxonkit lineage beam_tax.txt | tee ./domain_tax/beam_tax_lin.txt
parse JSON from foldseek search server
https://search.foldseek.com/search
- PDB database
- Swiss-Prot database (AI-predicted structures)
- OMG_Prot50 (Proteins are transcribed from the Open MetaGenomic.)
- malidup
- malisam
PDB database
download
- https://files.rcsb.org/pub/pdb/data/structures/all/pdb/
- save this website as PDB - FTP Archive over HTTP.html
- extract pdbXXXX.ent.gz list
from bs4 import BeautifulSoup
# how many ent.gz file
with open('PDB - FTP Archive over HTTP.html', 'r') as file:
html_content = file.read()
soup = BeautifulSoup(html_content, 'lxml')
text = soup.get_text('\n', '\n\n')
lines = text.split('\n')
PDB_id_list = []
for line in lines:
if 'ent.gz' in line:
PDB_id_list.append(line.split(".")[0][4:] #.split("pdb")[1])
# PDB_id_list[:5]
# ['100d', '101d', '101m', '102d', '102l']
It's not okay to extract id by
.split("pdb"). Because pdb might be also the part of the id in some special cases, for example, pdb1pdb.ent.gz.
- parallel downloads
# split into 44 entry_i.txt file
length = 5000
count = len(df)//length
for i in range(43):
subdf = df.iloc[i*5000:i*5000+5000]
sublist =subdf["id"].tolist()
string = ",".join(sublist)
text = open('groups/entry_'+ str(i)+'.txt', 'w')
text.write(string)
text.close()
final_sublist = df.iloc[43*5000:43*5000+5000]["id"].tolist()
finalstring = ",".join(final_sublist)
finaltext = open('groups/entry_'+ str(43)+'.txt', 'w')
finaltext.write(finalstring)
finaltext.close()
Obatin the batch-download script batch_download.sh from Batch Downloads with Shell Script
#!/bin/bash
cd /(your path)
for line in $(cat ./../groups/entry_list.txt); do
./../batch_download.sh -f ./../groups/${line} -p &
done
where entry_list.txt has entry_i.txt line by line.
$ bash parallel_download.sh > download.log
Failed download is common when Shell script downloads large files simutaneously. Therefore, it is important to check whether the script has downloaded the complete and correct ent.gz files. For example, by
gunzip *gz, the wrong .gz files will outputunzip error. And then, this wrong .gz file should be removed and we need to download it again.
desciption
At this moment, I download 218,546 PDB entries from PDB database.
Swiss-Prot database
download
UniProt provides the reviewed Swiss-Prot database.
Unipressed API client
description
In UniProt, Swiss-Prot has 571,864 entries with its corresponding fasta file. 549,724 entries have 3D struture. And most (513,805) of these structure are from AlphaFold prediction.

OMG_Prot50 database
download
huggingface.co/datasets/tattabio/OMG_prot50
description
The
OMG_prot50dataset is a protein-only dataset, created by clustering the Open MetaGenomic dataset (OMG) at 50% sequence identity. MMseqs2 linclust (Steinegger and Söding 2018) was used to cluster all 4.2B protein sequences from the OMG dataset, resulting in 207M protein sequences. Sequences were clustered at 50% sequence id and 90% sequence coverage, and singleton clusters were removed.
malidup
http://prodata.swmed.edu/malidup/
malisam
http://prodata.swmed.edu/malisam/
Transporter Classification Database (TCDB)
The Transporter Classification Database (TCDB) is specialized with respect to curated information about the functions and evolution of transporters from all domains of life [Saier et al., 2021].
sequence alignment via BLASTp with using makeblastdb for custom database
module load blast/2.11.0+
makeblastdb -in ./targetDB/target30.fasta -input_type fasta -dbtype prot -out ./targetDB_blast/targetDB.blast
blastp -query q92508.fasta -db ./targetDB_blast/targetDB.blast -outfmt 5 -out query_blastp_target30_fmt5.xml
tree
.
├── q92508.fasta
├── query_blastp_target30_fmt5.xml
├── query_blastp_target30.out
├── targetDB
│ └── target30.fasta
└── targetDB_blast
├── targetDB.blast.pdb
├── targetDB.blast.phr
├── targetDB.blast.pin
├── targetDB.blast.pot
├── targetDB.blast.psq
├── targetDB.blast.ptf
└── targetDB.blast.pto
- paese
from Bio.Blast import NCBIXML
with open("./query_blastp_target30_fmt5.xml") as f:
records = NCBIXML.parse(f)
for record in records:
for alignment in record.alignments:
print("Target:", alignment.hit_def)
for hsp in alignment.hsps:
print(" Score:", hsp.score)
print(" E-value:", hsp.expect)
print(" Query seq:", hsp.query)
print(" Sbjct seq:", hsp.sbjct)
print(" Match:", hsp.match)
Assessment
TM-score
Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004 Dec 1;57(4):702-10. doi: 10.1002/prot.20264. Erratum in: Proteins. 2007 Sep 1;68(4):1020. PMID: 15476259.

E-value
how randomly you find this structure. How likely you find this match just by chance.
Prob

pLDDT
"predicted Local Distance Difference Test" is a per-residue measure of local confidence. It is scaled from 0 to 100, with higher scores indicating higher confidence and usually a more accurate prediction. ref
PAE
Predicted Aligned Error (PAE) is a measure of how confident AlphaFold2 is in the relative position of two residues within the predicted structure. PAE is defined as the expected positional error at residue X, measured in Ångströms (Å), if the predicted and actual structures were aligned on residue Y.ref
fasta to mutated fasta (python code)
import copy
mutations = ["K114Q", "R196I", "Y20F", "K8Q"]
fa_header = open("./xxxx.fasta", "r").read().split("\n")[0]
fa_seq = open("./xxxx.fasta", "r").read().split("\n")[1]
new_seq = copy.copy(fa_seq)
for mutation in mutations:
orig = mutation[0]
idx = int(mutation[1:-1])
mut = mutation[-1]
seq_list = list(new_seq)
if seq_list[idx-1] == orig:
seq_list[idx-1] = mut
else:
print("Check!")
new_seq = ''.join(seq_list)
with open('mutatedfa.fa', 'w') as f:
f.write(fa_header)
f.write("\n")
f.write(new_seq)
sequence similarity
import parasail
# 1
result = parasail.sw_trace_scan_16("asdf", "asdf", 11, 1, parasail.blosum62)
# 2
edlib.align(x, y, mode='HW', task='locations', k=max_dist + 1) # edit distance
foldseek usage (straightforward)
Installation: Conda installer (Linux and macOS)
conda install -c conda-forge -c bioconda foldseek
Customize Database
mkdir DB
foldseek createdb myDB_dir/ ./DB/myDB
# myDB_dir has lots of pdb files
foldseek all2all
foldseek easy-search ./DB/myDB ./DB/myDB result.m8 tmp --alignment-type 1 --format-output query,target,evalue,qtmscore,ttmscore,alntmscore,rmsd,prob
foldseek easy-search
usage: foldseek easy-search <i:PDB|mmCIF[.gz]> ... <i:PDB|mmCIF[.gz]>|<i:stdin> <i:targetFastaFile[.gz]>|<i:targetDB> <o:alignmentFile> <tmpDir> [options]
By Martin Steinegger <martin.steinegger@snu.ac.kr>
options: prefilter:
--comp-bias-corr INT Correct for locally biased amino acid composition (range 0-1) [1]
--comp-bias-corr-scale FLOAT Correct for locally biased amino acid composition (range 0-1) [1.000]
--seed-sub-mat TWIN Substitution matrix file for k-mer generation [aa:3di.out,nucl:3di.out]
-s FLOAT Sensitivity: 1.0 faster; 4.0 fast; 7.5 sensitive [9.500]
-k INT k-mer length (0: automatically set to optimum) [6]
--target-search-mode INT target search mode (0: regular k-mer, 1: similar k-mer) [0]
--k-score TWIN k-mer threshold for generating similar k-mer lists [seq:2147483647,prof:2147483647]
--max-seqs INT Maximum results per query sequence allowed to pass the prefilter (affects sensitivity) [1000]
--split INT Split input into N equally distributed chunks. 0: set the best split automatically [0]
--split-mode INT 0: split target db; 1: split query db; 2: auto, depending on main memory [2]
--split-memory-limit BYTE Set max memory per split. E.g. 800B, 5K, 10M, 1G. Default (0) to all available system memory [0]
--diag-score BOOL Use ungapped diagonal scoring during prefilter [1]
--exact-kmer-matching INT Extract only exact k-mers for matching (range 0-1) [0]
--mask INT Mask sequences in k-mer stage: 0: w/o low complexity masking, 1: with low complexity masking [0]
--mask-prob FLOAT Mask sequences is probablity is above threshold [1.000]
--mask-lower-case INT Lowercase letters will be excluded from k-mer search 0: include region, 1: exclude region [1]
--min-ungapped-score INT Accept only matches with ungapped alignment score above threshold [30]
--spaced-kmer-mode INT 0: use consecutive positions in k-mers; 1: use spaced k-mers [1]
--spaced-kmer-pattern STR User-specified spaced k-mer pattern []
--local-tmp STR Path where some of the temporary files will be created []
--exhaustive-search BOOL Turns on an exhaustive all vs all search by by passing the prefilter step [0]
align:
--min-seq-id FLOAT List matches above this sequence identity (for clustering) (range 0.0-1.0) [0.000]
-c FLOAT List matches above this fraction of aligned (covered) residues (see --cov-mode) [0.000]
--cov-mode INT 0: coverage of query and target
1: coverage of target
2: coverage of query
3: target seq. length has to be at least x% of query length
4: query seq. length has to be at least x% of target length
5: short seq. needs to be at least x% of the other seq. length [0]
--max-rejected INT Maximum rejected alignments before alignment calculation for a query is stopped [2147483647]
--max-accept INT Maximum accepted alignments before alignment calculation for a query is stopped [2147483647]
-a BOOL Add backtrace string (convert to alignments with mmseqs convertalis module) [0]
--sort-by-structure-bits INT sort by bits*sqrt(alnlddt*alntmscore) [1]
--alignment-mode INT How to compute the alignment:
0: automatic
1: only score and end_pos
2: also start_pos and cov
3: also seq.id [3]
--alignment-output-mode INT How to compute the alignment:
0: automatic
1: only score and end_pos
2: also start_pos and cov
3: also seq.id
4: only ungapped alignment
5: score only (output) cluster format [0]
-e DOUBLE List matches below this E-value (range 0.0-inf) [1.000E+01]
--min-aln-len INT Minimum alignment length (range 0-INT_MAX) [0]
--seq-id-mode INT 0: alignment length 1: shorter, 2: longer sequence [0]
--alt-ali INT Show up to this many alternative alignments [0]
--gap-open TWIN Gap open cost [aa:10,nucl:10]
--gap-extend TWIN Gap extension cost [aa:1,nucl:1]
profile:
--num-iterations INT Number of iterative profile search iterations [1]
misc:
--tmscore-threshold FLOAT accept alignments with a tmsore > thr [0.0,1.0] [0.000]
--tmalign-hit-order INT order hits by 0: (qTM+tTM)/2, 1: qTM, 2: tTM, 3: min(qTM,tTM) 4: max(qTM,tTM) [0]
--tmalign-fast INT turn on fast search in TM-align [1]
--lddt-threshold FLOAT accept alignments with a lddt > thr [0.0,1.0] [0.000]
--alignment-type INT How to compute the alignment:
0: 3di alignment
1: TM alignment
2: 3Di+AA [2]
--exact-tmscore INT turn on fast exact TMscore (slow), default is approximate [0]
--prefilter-mode INT prefilter mode: 0: kmer/ungapped 1: ungapped, 2: nofilter [0]
--cluster-search INT first find representative then align all cluster members [0]
--mask-bfactor-threshold FLOAT mask residues for seeding if b-factor < thr [0,100] [0.000]
--input-format INT Format of input structures:
0: Auto-detect by extension
1: PDB
2: mmCIF
3: mmJSON
4: ChemComp
5: Foldcomp [0]
--file-include STR Include file names based on this regex [.*]
--file-exclude STR Exclude file names based on this regex [^$]
--format-mode INT Output format:
0: BLAST-TAB
1: SAM
2: BLAST-TAB + query/db length
3: Pretty HTML
4: BLAST-TAB + column headers
5: Calpha only PDB super-posed to query
BLAST-TAB (0) and BLAST-TAB + column headers (4)support custom output formats (--format-output)
(5) Superposed PDB files (Calpha only) [0]
--format-output STR Choose comma separated list of output columns from: query,target,evalue,gapopen,pident,fident,nident,qstart,qend,qlen
tstart,tend,tlen,alnlen,raw,bits,cigar,qseq,tseq,qheader,theader,qaln,taln,mismatch,qcov,tcov
qset,qsetid,tset,tsetid,taxid,taxname,taxlineage,
lddt,lddtfull,qca,tca,t,u,qtmscore,ttmscore,alntmscore,rmsd,prob
complexqtmscore,complexttmscore,complexu,complext,complexassignid
[query,target,fident,alnlen,mismatch,gapopen,qstart,qend,tstart,tend,evalue,bits]
--greedy-best-hits BOOL Choose the best hits greedily to cover the query [0]
common:
--db-load-mode INT Database preload mode 0: auto, 1: fread, 2: mmap, 3: mmap+touch [0]
--threads INT Number of CPU-cores used (all by default) [40]
-v INT Verbosity level: 0: quiet, 1: +errors, 2: +warnings, 3: +info [3]
--sub-mat TWIN Substitution matrix file [aa:3di.out,nucl:3di.out]
--max-seq-len INT Maximum sequence length [65535]
--compressed INT Write compressed output [0]
--remove-tmp-files BOOL Delete temporary files [1]
--mpi-runner STR Use MPI on compute cluster with this MPI command (e.g. "mpirun -np 42") []
--force-reuse BOOL Reuse tmp filse in tmp/latest folder ignoring parameters and version changes [0]
--prostt5-model STR Path to ProstT5 model []
expert:
--zdrop INT Maximal allowed difference between score values before alignment is truncated (nucleotide alignment only) [40]
--taxon-list STR Taxonomy ID, possibly multiple values separated by ',' []
--chain-name-mode INT Add chain to name:
0: auto
1: always add
[0]
--write-mapping INT write _mapping file containing mapping from internal id to taxonomic identifier [0]
--coord-store-mode INT Coordinate storage mode:
1: C-alpha as float
2: C-alpha as difference (uint16_t) [2]
--write-lookup INT write .lookup file containing mapping from internal id, fasta id and file number [1]
--db-output BOOL Return a result DB instead of a text file [0]
examples:
# Search a single/multiple PDB file against a set of PDB files
foldseek easy-search examples/d1asha_ examples/ result.m8 tmp
# Format output differently
foldseek easy-search examples/d1asha_ examples/ result.m8 tmp --format-output query,target,qstart,tstart,cigar
# Align with TMalign (global)
foldseek easy-search examples/d1asha_ examples/ result.m8 tmp --alignment-type 1
# Skip prefilter and perform an exhaustive alignment (slower but more sensitive)
foldseek easy-search examples/d1asha_ examples/ result.m8 tmp --exhaustive-search 1
usage history
#!/bin/bash
source ~/miniconda3/etc/profile.d/conda.sh
conda activate foldseek
pdb_DB="/(...)/lihuilin/myfoldseek/PDB_db_dir/myPDBdb"
afdb_DB="/(...)/lihuilin/myfoldseek/AFDB_db_dir/myAFDBdb"
query_pdb="/(...)/lihuilin/query/xxx.pdb"
cd /(...)/lihuilin/myfoldseek
mkdir -p xxx_all
cd xxx_all
# foldseek easy-search $query_pdb $afdb_DB xxx_mut_afdb_tms_thr_abo_0_4_result.m8 tmp --tmscore-threshold 0.4 --alignment-type 1 --format-output query,target,evalue,pident,fident,bits,tseq,qtmscore,ttmscore,alntmscore,rmsd,prob
# foldseek easy-search $query_pdb $pdb_DB xxx_mut_pdbdb_tms_thr_abo_0_4_result.m8 tmp --tmscore-threshold 0.4 --alignment-type 1 --format-output query,target,evalue,pident,fident,bits,tseq,qtmscore,ttmscore,alntmscore,rmsd,prob
afdb_res_m8="/(...)/lihuilin/myfoldseek/xxx_mut_afdb_tms_thr_abo_0_4_result.m8"
pdb_res_m8="/(...)/lihuilin/myfoldseek/xxx_mut_pdbdb_tms_thr_abo_0_4_result.m8"
PDBdb_dir="/(...)/lihuilin/DB/pure_PDB_DB_by_Chain/"
AFDBdb_dir="/(...)/lihuilin/DB/AFDB_pLDDT_70"
afdb_res_dir="/(...)/lihuilin/myfoldseek/xxx_mut_afdb_tms_thr_abo_0_4_result_dir"
pdb_res_dir="/(...)/lihuilin/myfoldseek/xxx_mut_pdbdb_tms_thr_abo_0_4_result_dir"
mkdir -p $afdb_res_dir
mkdir -p $pdb_res_dir
# for afdb
while read -r line; do
ID=$(echo "$line" | awk '{print $2}')
inp_fp="$AFDBdb_dir/$ID.pdb"
oup_fp="$afdb_res_dir/$ID.pdb"
cp "$inp_fp" "$oup_fp"
done < $afdb_res_m8
# for pdb
while read -r line; do
ID=$(echo "$line" | awk '{print $2}')
inp_fp="$PDBdb_dir$ID.pdb"
oup_fp="$pdb_res_dir/$ID.pdb"
cp "$inp_fp" "$oup_fp"
done < $pdb_res_m8
MMseqs2 usage (straightforward)
usage history
#!/bin/bash
cd /(...)/lihuilin/MMseqs2/
cd build
make
make install
export PATH=$(pwd)/bin/:$PATH
# echo "============================================== COMPILE ENVIRONMENT ========================================================="
cd /(...)/lihuilin/myMMseqs2
mmseqs easy-search /(...)/lihuilin/query/xxx_mut.fa /(...)/lihuilin/DB/PDB_UPSP_fasta_DB/faDB.fasta xxx_mut_mmseqs_id_0_2_c_0_7.m8 tmp --min-seq-id 0.2 -c 0.7 --format-output query,target,fident,evalue,bits,qheader,theader,tseq
# echo "============================================== .pdb file of these similar sequence ========================================================="
result_file="/(...)/lihuilin/myMMseqs2/xxx_mut_mmseqs_id_0_2_c_0_7.m8"
PDBdb_dir="/(...)/lihuilin/DB/pure_PDB_DB_by_Chain/"
AFDBdb_dir="/(...)/lihuilin/DB/AFDB_pLDDT_70"
output_dir="/(...)/lihuilin/myMMseqs2/xxx_mut_mmseqs_id_0_2_c_0_7_pdb_dir"
mkdir -p "$output_dir"
AFDB_pLDDT_70_log="/(...)/lihuilin/DB/AFDB_pLDDT_70.log"
pure_PDB_DB_by_Chain_log="/(...)/lihuilin/DB/pure_PDB_DB_by_Chain.log"
while read -r line; do
ID=$(echo "$line" | awk '{print $2}')
echo "$ID"
if grep -q "$ID.pdb" $pure_PDB_DB_by_Chain_log; then
inp_fp="$PDBdb_dir$ID.pdb"
oup_fp="$output_dir/$ID.pdb"
cp "$inp_fp" "$oup_fp"
elif grep -q "AF-$ID.pdb" $AFDB_pLDDT_70_log; then
inp_fp="$AFDBdb_dir/AF-$ID.pdb"
oup_fp="$output_dir/AF-$ID.pdb"
cp "$inp_fp" "$oup_fp"
else
echo "didnt download $ID structure"
fi
done < $result_file
multiple seqs search
#!/bin/bash
cd /(...)/lihuilin/MMseqs2/
cd build
make
make install
export PATH=$(pwd)/bin/:$PATH
windowsfa="/(...)/lihuilin/query/xxx_mut_windows.fa"
m8fp="/(...)/lihuilin/myMMseqs2/xxx_mut_windows_mmseqs.m8"
mmseqs easy-search $windowsfa /(...)/lihuilin/DB/PDB_UPSP_fasta_DB/faDB.fasta $m8fp tmp --format-output query,target,fident,evalue,nident,alnlen,mismatch,qstart,qend,tstart,tend,bits,qheader,theader,tseq
localcolabfold usage (straightforward)
localcolabfold is ColabFold for protein structure prediction on local PC.
One protein prediction in command line
- msa only
conda activate /(...)/lihuilin/mycolabfold/localcolabfold/colabfold-conda
module load cuda/12.0
colabfold_batch fafile outdir --msa-only
- GPU prediction
conda activate /(...)/lihuilin/mycolabfold/localcolabfold/colabfold-conda
module load cuda/12.0
module load gcc/9.4.0
colabfold_batch fafile outdir --num-relax 1
Multiple proteins prediction in slurm scrpits
- msa only: msaonly.sh
#!/bin/bash
source ~/miniconda3/etc/profile.d/conda.sh
conda activate /(...)/lihuilin/mycolabfold/localcolabfold/colabfold-conda
module load cuda/12.0
cd /(...)/fasta_files
INPUT_FILES=($(ls *.fasta))
INPUT=${INPUT_FILES[$SLURM_ARRAY_TASK_ID-1]}
OUTPUT="${INPUT%.*}_out"
colabfold_batch $INPUT $OUTPUT --msa-only
bash msaonly.sh
- GPU prediction
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH -p a100-40g
#SBATCH --gres=gpu:1
#SBATCH --mem=80G
#SBATCH -J JobName
#SBATCH -a 1-4
module load cuda/12.0
module load gcc/9.4.0
source ~/miniconda3/etc/profile.d/conda.sh
conda activate /storage/(...)/lihuilin/mycolabfold/localcolabfold/colabfold-conda
cd /(...)/fasta_files
INPUT_FILES=($(ls *.fasta))
INPUT=${INPUT_FILES[$SLURM_ARRAY_TASK_ID-1]}
OUTPUT="${INPUT%.*}_out"
colabfold_batch $INPUT $OUTPUT --num-relax 1
Encoding Protein Sequences into Integer Representations Using K-mer Binary Mapping
seq="MKTLGEFIVEKQH",k=4.- Kmers:
['MKTL', 'KTLG', 'TLGE', 'LGEF', 'GEFI', 'EFIV', 'FIVE', 'IVEK', 'VEKQ', 'EKQH'] - Encodings:
[5057, 15385, 49556, 6470, 37986, 17954, 25124, 8771, 9268, 17226] - Check:
| Kmer | K-mer Binary Mapping | Encoding | Check |
|---|---|---|---|
| "MKTL" | 0001 0011 1100 0001 | 5057 | ✔️ |
| "KTLG" | 0011 1100 0001 1001 | 15385 | ✔️ |
| "TLGE" | 1100 0001 1001 0100 | 49556 | ✔️ |
| "LGEF" | 0001 1001 0100 0110 | 6470 | ✔️ |
| "GEFI" | 1001 0100 0110 0010 | 37986 | ✔️ |
| "EFIV" | 0100 0110 0010 0010 | 17954 | ✔️ |
| "FIVE" | 0110 0010 0010 0100 | 25124 | ✔️ |
| "IVEK" | 0010 0010 0100 0011 | 8771 | ✔️ |
| "VEKQ" | 0010 0100 0011 0100 | 9268 | ✔️ |
| "EKQH" | 0100 0011 0100 1010 | 17226 | ✔️ |
- Code:
parasail in C usage (straightforward)
I want to use its Tracebacks functions, however, this function has not been involved in Python version. Therefore, I am exploring its C usage.
installation ("building from a git clone")
straightforward steps:
cd /storage/lihuilin/git clone https://github.com/jeffdaily/parasailcd parasailautoreconf -fi
output
libtoolize: putting auxiliary files in AC_CONFIG_AUX_DIR, 'build-aux'.
libtoolize: copying file 'build-aux/ltmain.sh'
libtoolize: putting macros in AC_CONFIG_MACRO_DIRS, 'm4'.
libtoolize: copying file 'm4/libtool.m4'
libtoolize: copying file 'm4/ltoptions.m4'
libtoolize: copying file 'm4/ltsugar.m4'
libtoolize: copying file 'm4/ltversion.m4'
libtoolize: copying file 'm4/lt~obsolete.m4'
configure.ac:109: installing 'build-aux/compile'
configure.ac:78: installing 'build-aux/config.guess'
configure.ac:78: installing 'build-aux/config.sub'
configure.ac:70: installing 'build-aux/install-sh'
configure.ac:70: installing 'build-aux/missing'
Makefile.am: installing 'build-aux/depcomp'
parallel-tests: installing 'build-aux/test-driver'
mkdir -p /storage/lihuilin/parasail/local./configure --prefix=/storage/lihuilin/parasail/local/parasail
output
... ...
-=-=-=-=-=-=-=-=-=-= Configuration Complete =-=-=-=-=-=-=-=-=-=-=-
Configuration summary :
parasail version : .................... 2.6.2
Host CPU : ............................ x86_64
Host Vendor : ......................... pc
Host OS : ............................. linux-gnu
Toolchain :
CC : .................................. gcc (gnu, 8.3.1)
CXX : ................................. g++ (gnu, 8.3.1)
Flags :
CFLAGS : .............................. -g -O2
CXXFLAGS : ............................ -g -O2
CPPFLAGS : ............................
LDFLAGS : .............................
LIBS : ................................
Intrinsics :
SSE2 : ................................ auto (yes)
SSE2_CFLAGS : .........................
SSE4.1 : .............................. auto (yes)
SSE41_CFLAGS : ........................ -msse4.1
AVX2 : ................................ auto (yes)
AVX2_CFLAGS : ......................... -mavx2
AVX512 : .............................. auto (yes)
AVX512F_CFLAGS : ...................... -mavx512f
AVX512BW_CFLAGS : ..................... -mavx512bw
AVX512VBMI_CFLAGS : ................... -mavx512vbmi
Altivec : ............................. auto (no)
ALTIVEC_CFLAGS : ...................... not supported
ARM NEON : ............................ auto (no)
NEON_CFLAGS : ......................... not supported
EXTRA_NEON_CFLAGS : ................... -fopenmp-simd -DSIMDE_ENABLE_OPENMP
Dependencies :
OPENMP_CFLAGS : ....................... -fopenmp
OPENMP_CXXFLAGS : ..................... -fopenmp
CLOCK_LIBS : ..........................
MATH_LIBS : ........................... -lm
Z_CFLAGS : ............................
Z_LIBS : .............................. -lz
Installation directories :
Program directory : ................... /storage/lihuilin/parasail/local/parasail/bin
Library directory : ................... /storage/lihuilin/parasail/local/parasail/lib
Include directory : ................... /storage/lihuilin/parasail/local/parasail/include
Pkgconfig directory : ................. /storage/lihuilin/parasail/local/parasail/lib/pkgconfig
Compiling some other packages against parasail may require
the addition of '/storage/lihuilin/parasail/local/parasail/lib/pkgconfig' to the
PKG_CONFIG_PATH environment variable.
make
output
make all-am
make[1]: Entering directory '/storage/lihuilin/parasail'
... ...
make[1]: Leaving directory '/storage/lihuilin/parasail'
make install
output
make[1]: Entering directory '/storage/lihuilin/parasail'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/lib'
/bin/sh ./libtool --mode=install /usr/bin/install -c libparasail.la '/storage/lihuilin/parasail/local/parasail/lib'
libtool: install: /usr/bin/install -c .libs/libparasail.so.8.1.2 /storage/lihuilin/parasail/local/parasail/lib/libparasail.so.8.1.2
libtool: install: (cd /storage/lihuilin/parasail/local/parasail/lib && { ln -s -f libparasail.so.8.1.2 libparasail.so.8 || { rm -f libparasail.so.8 && ln -s libparasail.so.8.1.2 libparasail.so.8; }; })
libtool: install: (cd /storage/lihuilin/parasail/local/parasail/lib && { ln -s -f libparasail.so.8.1.2 libparasail.so || { rm -f libparasail.so && ln -s libparasail.so.8.1.2 libparasail.so; }; })
libtool: install: /usr/bin/install -c .libs/libparasail.lai /storage/lihuilin/parasail/local/parasail/lib/libparasail.la
libtool: install: /usr/bin/install -c .libs/libparasail.a /storage/lihuilin/parasail/local/parasail/lib/libparasail.a
libtool: install: chmod 644 /storage/lihuilin/parasail/local/parasail/lib/libparasail.a
libtool: install: ranlib /storage/lihuilin/parasail/local/parasail/lib/libparasail.a
libtool: finish: PATH="/home/lihuilin/miniconda3/condabin:/home/lihuilin/.local/bin:/home/lihuilin/bin:/opt/slurm/sbin:/opt/slurm/bin:/soft/modules/modules-4.7.0/bin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home/lihuilin/software/silent_tools:/sbin" ldconfig -n /storage/lihuilin/parasail/local/parasail/lib
----------------------------------------------------------------------
Libraries have been installed in:
/storage/lihuilin/parasail/local/parasail/lib
If you ever happen to want to link against installed libraries
in a given directory, LIBDIR, you must either use libtool, and
specify the full pathname of the library, or use the '-LLIBDIR'
flag during linking and do at least one of the following:
- add LIBDIR to the 'LD_LIBRARY_PATH' environment variable
during execution
- add LIBDIR to the 'LD_RUN_PATH' environment variable
during linking
- use the '-Wl,-rpath -Wl,LIBDIR' linker flag
- have your system administrator add LIBDIR to '/etc/ld.so.conf'
See any operating system documentation about shared libraries for
more information, such as the ld(1) and ld.so(8) manual pages.
----------------------------------------------------------------------
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/bin'
/bin/sh ./libtool --mode=install /usr/bin/install -c apps/parasail_aligner apps/parasail_stats '/storage/lihuilin/parasail/local/parasail/bin'
libtool: install: /usr/bin/install -c apps/.libs/parasail_aligner /storage/lihuilin/parasail/local/parasail/bin/parasail_aligner
libtool: install: /usr/bin/install -c apps/.libs/parasail_stats /storage/lihuilin/parasail/local/parasail/bin/parasail_stats
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/include'
/usr/bin/install -c -m 644 parasail.h '/storage/lihuilin/parasail/local/parasail/include'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/include'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/include/parasail/matrices'
/usr/bin/install -c -m 644 parasail/matrices/blosum100.h parasail/matrices/blosum30.h parasail/matrices/blosum35.h parasail/matrices/blosum40.h parasail/matrices/blosum45.h parasail/matrices/blosum50.h parasail/matrices/blosum55.h parasail/matrices/blosum60.h parasail/matrices/blosum62.h parasail/matrices/blosum65.h parasail/matrices/blosum70.h parasail/matrices/blosum75.h parasail/matrices/blosum80.h parasail/matrices/blosum85.h parasail/matrices/blosum90.h parasail/matrices/blosum_map.h parasail/matrices/blosumn.h parasail/matrices/dnafull.h parasail/matrices/nuc44.h parasail/matrices/pam10.h parasail/matrices/pam100.h parasail/matrices/pam110.h parasail/matrices/pam120.h parasail/matrices/pam130.h parasail/matrices/pam140.h parasail/matrices/pam150.h parasail/matrices/pam160.h parasail/matrices/pam170.h parasail/matrices/pam180.h parasail/matrices/pam190.h parasail/matrices/pam20.h parasail/matrices/pam200.h parasail/matrices/pam210.h parasail/matrices/pam220.h parasail/matrices/pam230.h parasail/matrices/pam240.h parasail/matrices/pam250.h parasail/matrices/pam260.h parasail/matrices/pam270.h parasail/matrices/pam280.h '/storage/lihuilin/parasail/local/parasail/include/parasail/matrices'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/include/parasail/matrices'
/usr/bin/install -c -m 644 parasail/matrices/pam290.h parasail/matrices/pam30.h parasail/matrices/pam300.h parasail/matrices/pam310.h parasail/matrices/pam320.h parasail/matrices/pam330.h parasail/matrices/pam340.h parasail/matrices/pam350.h parasail/matrices/pam360.h parasail/matrices/pam370.h parasail/matrices/pam380.h parasail/matrices/pam390.h parasail/matrices/pam40.h parasail/matrices/pam400.h parasail/matrices/pam410.h parasail/matrices/pam420.h parasail/matrices/pam430.h parasail/matrices/pam440.h parasail/matrices/pam450.h parasail/matrices/pam460.h parasail/matrices/pam470.h parasail/matrices/pam480.h parasail/matrices/pam490.h parasail/matrices/pam50.h parasail/matrices/pam500.h parasail/matrices/pam60.h parasail/matrices/pam70.h parasail/matrices/pam80.h parasail/matrices/pam90.h parasail/matrices/pam_map.h '/storage/lihuilin/parasail/local/parasail/include/parasail/matrices'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/include/parasail'
/usr/bin/install -c -m 644 parasail/cpuid.h parasail/io.h parasail/function_lookup.h parasail/matrix_lookup.h '/storage/lihuilin/parasail/local/parasail/include/parasail'
/usr/bin/mkdir -p '/storage/lihuilin/parasail/local/parasail/lib/pkgconfig'
/usr/bin/install -c -m 644 parasail-1.pc '/storage/lihuilin/parasail/local/parasail/lib/pkgconfig'
make[1]: Leaving directory '/storage/lihuilin/parasail'
usage
cd /storage/lihuilin/parasail/local/parasail/bin
.
├── myseqs.fasta
├── parasail_aligner
└── parasail_stats
myseqs.fasta is
>sequence1
CYAMYGSSTHLVLTLGDGVDGFTLDTNLGEFILTHPNLRIPPQKAIYSINEGPCDKKSPNGKLRLLYEAFPMAFLMEQAGGKAVNDRGERILDLVPSHIHDKSSIWLGSSGEIDKFLDHIGKSQ
>sequence2
IKFPNGVQKYIKFCQEEDKSTNRPYTSRYIGSLVADFHRNLLKGGIYLYPSTASHPDGKLRLLYECNPMAFLAEQAGGKASDGKERILDIIPETLHQRRSFFVGNDHMVEDVERFIREFPDA
- In terminal,
./parasail_aligner -f myseqs.fasta -O EMBOSS -a sw_trace_scan_16
sequence2 50 PSTASHPDGKLRLLYECNPMAFLAEQAGGKA-SDGKERILDIIPETLHQR 98
|.....|:||||||||..|||||.||||||| :|..|||||::|..:|.:
sequence1 53 PCDKKSPNGKLRLLYEAFPMAFLMEQAGGKAVNDRGERILDLVPSHIHDK 102
sequence2 99 RSFFVGNDHMVEDVERFI 116
.|.::|:. .::::|:
sequence1 103 SSIWLGSS---GEIDKFL 117
Length: 68
Identity: 33/68 (48.5%)
Similarity: 47/68 (69.1%)
Gaps: 4/68 ( 5.9%)
Score: 162
./parasail_aligner -h
usage: parasail_aligner [-a funcname] [-c cutoff] [-x] [-e gap_extend] [-o gap_open] [-m matrix] [-t threads] [-d] [-M match] [-X mismatch] [-k band size (for nw_banded)] [-l AOL] [-s SIM] [-i OS] [-v] [-V] -f file [-q query_file] [-g output_file] [-O output_format {EMBOSS,SAM,SAMH,SSW}] [-b batch_size] [-r memory_budget] [-C] [-A alphabet_aliases]
Defaults:
funcname: sw_stats_striped_16
cutoff: 7, must be >= 1, exact match length cutoff
-x: if present, don't use suffix array filter
gap_extend: 1, must be >= 0
gap_open: 10, must be >= 0
matrix: blosum62
-d: if present, assume DNA alphabet ACGT
match: 1, must be >= 0
mismatch: 0, must be >= 0
threads: system-specific default, must be >= 1
AOL: 80, must be 0 <= AOL <= 100, percent alignment length
SIM: 40, must be 0 <= SIM <= 100, percent exact matches
OS: 30, must be 0 <= OS <= 100, percent optimal score
over self score
-v: verbose output, report input parameters and timing
-V: verbose memory output, report memory use
file: no default, must be in FASTA format
query_file: no default, must be in FASTA format
output_file: parasail.csv
output_format: no default, must be one of {EMBOSS,SAM,SAMH,SSW}
batch_size: 0 (calculate based on memory budget),
how many alignments before writing output
memory_budget: 2GB or half available from system query (100.970 GB)
-C: if present, use case sensitive alignments, matrices, etc.
alphabet_aliases: traceback will treat these pairs of characters as matches,
for example, 'TU' for one pair, or multiple pairs as 'XYab'
Biopython
SeqIO.parse()
from Bio import SeqIO
with open("fa.fasta") as fa:
for record in SeqIO.parse(fa, "fasta"):
ID = record.id
seq = record.seq
description = recrod.description
from Bio import PDB
if __name__=="__main__":
for pdbfile_path in glob.glob("/path/DB/pure_PDB/*.pdb"):
name = pdbfile_path.split("\\")[-1].split(".")[0]
print(name, end=" ")
pdb = PDB.PDBParser().get_structure(name, pdbfile_path)
pdb_io = PDB.PDBIO()
pdb_chains = pdb.get_chains()
for chain in pdb_chains:
pdb_io.set_structure(chain)
pdb_io.save("/path/DB/pure_PDB_DB_by_Chain/" + pdb.get_id() + "_" + chain.get_id() + ".pdb")
print('-- Done')
from Bio import SeqIO
def pdb2fasta(pdb_file, fasta_file, HEADER=None):
with open(pdb_file, 'r') as pdb_file:
for record in SeqIO.parse(pdb_file, 'pdb-atom'):
if '?' in record.id:
header = HEADER
else:
header = record.id
with open(fasta_file, 'w') as fasta_file:
fasta_file.write('>' + header+'\n')
fasta_file.write(str(record.seq))
return record.seq
from Bio import SeqIO
with open('fa.fasta') as fasta_file:
identifiers, seq = [], []
for seq_record in SeqIO.parse(fasta_file, 'fasta'):
identifiers.append(seq_record.id)
seq.append("".join(seq_record.seq))
fa_df = pd.DataFrame()
fa_df["seq"] = seq
fa_df["id"] = identifiers
remove DNA/RNA and O from pdb format
from Bio import PDB
class rm_dna_rna_O(PDB.Select):
def accept_residue(self, res):
if (len(res.get_resname()) <3) or (res.id[0] != " "):
return False
else:
return True
if __name__=="__main__":
for pdbfile_path in glob.glob("/path/PDBDB/*.pdb"):
print(pdbfile_path, end=" ")
name = pdbfile_path.split("/")[-1]
pdb = PDB.PDBParser().get_structure(name, pdbfile_path)
pdb_io = PDB.PDBIO()
pdb_io.set_structure(pdb)
pdb_io.save("/path/PDBDB_no_drna/"+name, rm_dna_rna_O())
print('-- Done')
remove HETATOM from pdb format
from Bio import PDB
pdb = PDB.PDBParser().get_structure("2gq1", "FBP/2gq1.pdb")
class ResSelect(PDB.Select):
def accept_residue(self, res):
if res.id[0] != " ": #>= start_res and res.id[1] <= end_res and res.parent.id == chain_id:
return False
else:
return True
pdb_io = PDB.PDBIO()
pdb_io.set_structure(pdb)
pdb_io.save("FBP/2gq1_no_HETATOM.pdb", ResSelect())
import os, gzip, shutil
import glob
import numpy as np
import pandas as pd
from Bio import SeqIO
# unzip all .gz files
start_dir = "./ftp.ensembl.org/pub/current_fasta/"
gz_count = []
destination_dir = "./fa_save/"
for root, dirs, files in os.walk(start_dir):
for gz_name in files:
gz_file = os.path.join(root, gz_name)
if "/pep/" in gz_file:
# print(gz_file)
gz_count.append(gz_file)
out_file = destination_dir + gz_name.split(".gz")[0]
with gzip.open(gz_file,"rb") as f_in, open(out_file,"wb") as f_out:
shutil.copyfileobj(f_in, f_out)
# read
with open("./fa_save/all_data.fa") as fasta_file:
identifiers = []
seq = []
for seq_record in SeqIO.parse(fasta_file, 'fasta'):
identifiers.append(seq_record.id)
seq.append("".join(seq_record.seq))
df_pro = pd.DataFrame()
df_pro["aa_seq"] = seq
df_pro["p_id"] = identifiers
# clear duplicates
df_pro_drop_dup = df_pro.drop_duplicates(subset='aa_seq')
use unipressed.UniprotkbClient analysis UniProt(Swiss-Prot) DB
We can reveal these information.
{'AFDB': 'hasAFDB',
'PDB': 'no',
'annotationScore': 1.0,
'comments': nan,
'entryAudit': "{'firstPublicDate': '1988-08-01', 'lastAnnotationUpdateDate': "
"'2022-05-25', 'lastSequenceUpdateDate': '1988-08-01', "
"'entryVersion': 36, 'sequenceVersion': 1}",
'entryType': 'UniProtKB reviewed (Swiss-Prot)',
'extraAttributes': "{'countByFeatureType': {'Chain': 1}, 'uniParcId': "
"'UPI000013C2E9'}",
'features': "[{'type': 'Chain', 'location': {'start': {'value': 1, "
"'modifier': 'EXACT'}, 'end': {'value': 79, 'modifier': "
"'EXACT'}}, 'description': 'Putative uncharacterized protein Z', "
"'featureId': 'PRO_0000066556'}]",
'geneLocations': nan,
'genes': nan,
'index': 860,
'keywords': "[{'id': 'KW-1185', 'category': 'Technical term', 'name': "
"'Reference proteome'}]",
'organism': "{'scientificName': 'Ovis aries', 'commonName': 'Sheep', "
"'taxonId': 9940, 'lineage': ['Eukaryota', 'Metazoa', 'Chordata', "
"'Craniata', 'Vertebrata', 'Euteleostomi', 'Mammalia', "
"'Eutheria', 'Laurasiatheria', 'Artiodactyla', 'Ruminantia', "
"'Pecora', 'Bovidae', 'Caprinae', 'Ovis']}",
'organismHosts': nan,
'primaryAccession': 'P08105',
'proteinDescription': "{'recommendedName': {'fullName': {'value': 'Putative "
"uncharacterized protein Z'}}}",
'proteinExistence': '4: Predicted',
'references': "[{'referenceNumber': 1, 'citation': {'id': '6193483', "
"'citationType': 'journal article', 'authors': ['Powell B.C.', "
"'Sleigh M.J.', 'Ward K.A.', 'Rogers G.E.'], "
"'citationCrossReferences': [{'database': 'PubMed', 'id': "
"'6193483'}, {'database': 'DOI', 'id': "
"'10.1093/nar/11.16.5327'}], 'title': 'Mammalian keratin gene "
'families: organisation of genes coding for the B2 high-sulphur '
"proteins of sheep wool.', 'publicationDate': '1983', "
"'journal': 'Nucleic Acids Res.', 'firstPage': '5327', "
"'lastPage': '5346', 'volume': '11'}, 'referencePositions': "
"['NUCLEOTIDE SEQUENCE [GENOMIC DNA]']}]",
'secondaryAccessions': nan,
'sequence': "{'value': "
"'MSSSLEITSFYSFIWTPHIGPLLFGIGLWFSMFKEPSHFCPCQHPHFVEVVIPCDSLSRSLRLRVIVLFLAIFFPLLNI', "
"'length': 79, 'molWeight': 9128, 'crc64': 'A663EB489F6290C3', "
"'md5': '80A5E20AFD024495655E6A9FA7B6166B'}",
'uniProtKBCrossReferences': "[{'database': 'EMBL', 'id': 'X01610', "
"'properties': [{'key': 'ProteinId', 'value': "
"'CAA25758.1'}, {'key': 'Status', 'value': '-'}, "
"{'key': 'MoleculeType', 'value': "
"'Genomic_DNA'}]}, {'database': 'PIR', 'id': "
"'S07912', 'properties': [{'key': 'EntryName', "
"'value': 'S07912'}]}, {'database': "
"'AlphaFoldDB', 'id': 'P08105', 'properties': "
"[{'key': 'Description', 'value': '-'}]}, "
"{'database': 'Proteomes', 'id': 'UP000002356', "
"'properties': [{'key': 'Component', 'value': "
"'Unplaced'}]}]",
'uniProtkbId': 'Z_SHEEP'}
from unipressed import UniprotkbClient
import pandas as pd
# Entries in Uniprot(Swiss-Prot)
uniprot_sp_entries_txt = open("/path/uniprot_sprot.fasta/SwissProt_entries.txt", "r")
uniprot_sp_entries = uniprot_sp_entries_txt.read().split('\n')
print(len(uniprot_sp_entries), uniprot_sp_entries[-3:])
# Split the whole entries in Uniprot(Swiss-Prot) into some chunks with the size of 1000
chunks = [uniprot_sp_entries[x:x+1000] for x in range(0, len(uniprot_sp_entries), 1000)]
print("We have ", len(chunks), " chunks, and the last chunk has ", len(chunks[-1])," entries.")
# analyze using unipressed
no_uniProtKBCrossReferences_list = []
no_uniProtKBCrossReferences_count = 0
for i in range(len(chunks)):
print(i, end=": ")
chunk = chunks[i]
db_dic = UniprotkbClient.fetch_many(chunk)
df = pd.DataFrame(db_dic)
print("chunk size is ", len(df), end="; ")
# no_uniProtKBCrossReferences list and count
no_uniProtKBCrossReferences_primaryAccession_list = df[df["uniProtKBCrossReferences"].isna()]["primaryAccession"].tolist()
no_uniProtKBCrossReferences_list.extend(no_uniProtKBCrossReferences_primaryAccession_list)
no_uniProtKBCrossReferences_count = no_uniProtKBCrossReferences_count + len(no_uniProtKBCrossReferences_primaryAccession_list)
print("no_uniProtKBCrossReferences_count is ", no_uniProtKBCrossReferences_count)
# has uniProtKBCrossReferences df
db_df = df[~df["uniProtKBCrossReferences"].isna()]
# has PDB structure or has AFDB structure
db_df.loc[:,["PDB"]] = db_df["uniProtKBCrossReferences"].apply(lambda x: "hasPDB" if "PDB" in pd.DataFrame(x)["database"].tolist() else "no")
db_df.loc[:,["AFDB"]] = db_df["uniProtKBCrossReferences"].apply(lambda x: "hasAFDB" if "AlphaFoldDB" in pd.DataFrame(x)["database"].tolist() else "no")
db_df.to_csv("/path/UniprotkbClient_results/db_df_"+str(i)+".csv")
# write no_uniProtKBCrossReferences_list to txt file
no_uniProtKBCrossReferences_file = open("/path/UniprotkbClient_results/no_uniProtKBCrossReferences_entries.txt", "w")
for line in no_uniProtKBCrossReferences_list:
no_uniProtKBCrossReferences_file.write(line+"\n")
no_uniProtKBCrossReferences_file.close()
DaliLite.V5 usage
http://ekhidna2.biocenter.helsinki.fi/dali/README.v5.html
../bin/dali.pl
--cd1 2nrmA # --cd1 <xxxxX> query structure identifier
--db pdb.list #--db <filename> list of target structure identifiers
--TITLE systematic
--dat1 ../DAT # path to directory containing query data [default: ./DAT/]
--dat2 ../DAT # path to directory containing target data [default: ./DAT/]
> systematic.stdout
2> systematic.stderr
dali name assignment
import itertools
import os, gzip, shutil
## roginal name 2 daliname
def generate_unique_names():
# Create a pool of characters: A-Z, 0-9
characters = 'abcdefghijklmnopqrstuvwxyz0123456789'
# Generate all combinations of 4 characters
unique_names = [''.join(comb) for comb in itertools.product(characters, repeat=4)]
return unique_names
unique_names = generate_unique_names()
orginalPDB_path = "./PDB/"
orginalPDB_fps = glob.glob(os.path.join(orginalPDB_path, "*.pdb"))
daliPDB_path = "./daliPDB/"
orig2dali_rows = []
for i in range(len(orginalPDB_fps)):
fp = orginalPDB_fps[i]
origname = fp.split("\\")[-1]
daliname = unique_names[i]
row = {"orginalPDB":origname, "daliPDB":daliname}
orig2dali_rows.append(row)
orig2dali_df = pd.DataFrame(orig2dali_rows)
orig2dali_df.to_csv("orig2dali_df.csv")
shutil.copyfile(fp, daliPDB_path + daliname + ".pdb")
import.pl
#!/bin/bash
cd /storage/shenhuaizhongLab/lihuilin/myDali/DaliLite.v5/piezo_ALL/daliPDB
module load blast/2.11.0+
mkdir -p "datdir"
for file in ./*.pdb; do
id=$(basename $file .pdb | head -c 4)
../../bin/import.pl --pdbfile $file --pdbid $id --dat datdir --clean
done
bash import.sh
dali.pl
- one2all, only Z-score
../bin/dali.pl -cd1 aaamA --db ./dat.txt --TITLE sysm --dat1 ./datdir --dat2 ./datdir
- one2one, with alignment
../bin/dali.pl --cd1 aaamA --cd2 aaa0A --dat1 ./datdir --dat2 ./datdir --title "output options" --outfmt "summary,alignments,equivalences,transrot" --clean 2> err
- one2all, with alignment
#!/bin/bash
cd /storage/shenhuaizhongLab/lihuilin/myDali/DaliLite.v5/piezo_ALL
module load blast/2.11.0+
DATDIR=/storage/shenhuaizhongLab/lihuilin/myDali/DaliLite.v5/piezo_ALL/datdir
REF=aaamA
for dat in "$DATDIR"/*.dat; do
id=$(basename "$dat" .dat)
if [ "$id" = "$REF" ]; then
continue
fi
jobdir="${REF}_vs_${id}"
echo "Aligning $REF vs $id → $jobdir"
mkdir -p "$jobdir"
(
cd "$jobdir" || exit 1
../../bin/dali.pl \
--cd1 "$REF" \
--cd2 "$id" \
--dat1 "$DATDIR" \
--dat2 "$DATDIR" \
--title "output options" \
--outfmt "summary,alignments,equivalences,transrot" \
--clean \
2> "err_${REF}_vs_${id}.log"
)
done
usage history
#!/bin/bash
module load blast/2.11.0+
# cd /(...)/lihuilin/myDali/DaliLite.v5/xxx_ALL/
# dat_dir="omg_xxx_dir"
# ls $dat_dir | perl -pe 's/\.dat//' > $dat_dir.list
# sys_dir="omg_xxx_sysm"
# mkdir -p "$sys_dir"
# cd $sys_dir
# ../../bin/dali.pl -cd1 0000A --db ../$dat_dir.list --TITLE sysm --dat1 ../$dat_dir --dat2 ../$dat_dir
for rep in aanwA bbydA bbx7A aafwA bbx4A bbyqA aabhA bbx5A bbx6A bbnuA
do
cd /(...)/lihuilin/myDali/DaliLite.v5/xxx_ALL/
dat_dir=$rep"_dat"
ls $dat_dir | perl -pe 's/\.dat//' > $dat_dir.list
sys_dir=$rep"_sysm"
mkdir -p "$sys_dir"
cd $sys_dir
../../bin/dali.pl -cd1 0000A --db ../$dat_dir.list --TITLE sysm --dat1 ../$dat_dir --dat2 ../$dat_dir
done
#../../bin/dali.pl -cd1 0000A --db ../xxxpdbR1.list --TITLE xxx_sysm --dat1 ../xxxDAT1 --dat2 ../xxxDAT1
#!/bin/bash
cd /storage/shenhuaizhongLab/lihuilin/myDali/DaliLite.v5/YYY_ALL
module load blast/2.11.0+
# mkdir -p "omg_fbp_dir"
# for file in ./*.pdb; do
# id=$(basename $file .pdb | head -c 4)
# ../bin/import.pl --pdbfile $file --pdbid $id --dat omg_fbp_dir --clean
# done
for rep in chunk1 chunk2 chunk3 chunk4 chunk5
do
dat_dir=$rep"_dat"
mkdir -p "$dat_dir"
../bin/import.pl --pdbfile ./0000A.pdb --pdbid 0000 --dat $dat_dir --clean
for file in $rep/*.pdb; do
id=$(basename $file .pdb | head -c 4)
../bin/import.pl --pdbfile $file --pdbid $id --dat $dat_dir --clean
done
done
.cif to .pdb
PyMol
File > Export Structure > Export Molecule > Save > change "save as type"
SeqIO.parse
from Bio import SeqIO
records = SeqIO.parse("input_file.cif", "cif-atom")
SeqIO.write(records, "output_file.pdb", "pdb-atom")
RCSB maxit
maxit -i file.cif -o 2
CD-HIT
from pycdhit import read_fasta, cd_hit, read_clstr, CDHIT
df_in = read_fasta("./up2now_piezo55.fasta")
cdhit = CDHIT(prog="cd-hit", path="~/tools/cd-hit-v4.8.1-2019-0228")
df_out, df_clstr = cdhit.set_options(c=0.9).cluster(df_in)
print(df_clstr)
parse Blastp XML2 file
NS = {"n": "http://www.ncbi.nlm.nih.gov"} # default namespace in your XML
def parse_blastxml2(path: str):
tree = ET.parse(path)
root = tree.getroot()
records = []
# Find each Search block
for search in root.findall(".//n:Results/n:search/n:Search", NS):
rec = {
# "query_id": search.findtext("n:query-id", default="", namespaces=NS),
"query_title": search.findtext("n:query-title", default="", namespaces=NS),
"query_len": int(search.findtext("n:query-len", default="0", namespaces=NS)),
"hits": []
}
for hit in search.findall("n:hits/n:Hit", NS):
descr = hit.find("n:description/n:HitDescr", NS)
taxid_text = descr.findtext("n:taxid", default="", namespaces=NS) if descr is not None else ""
taxid = int(taxid_text) if taxid_text.isdigit() else None
hit_obj = {
"num": int(hit.findtext("n:num", default="0", namespaces=NS)),
"accession": descr.findtext("n:accession", default="", namespaces=NS) if descr is not None else "",
"id": descr.findtext("n:id", default="", namespaces=NS) if descr is not None else "",
"title": descr.findtext("n:title", default="", namespaces=NS) if descr is not None else "",
"taxid": taxid,
"sciname": descr.findtext("n:sciname", default="", namespaces=NS) if descr is not None else "",
"len": int(hit.findtext("n:len", default="0", namespaces=NS)),
"hsps": []
}
for hsp in hit.findall("n:hsps/n:Hsp", NS):
hit_obj["hsps"].append({
"num": int(hsp.findtext("n:num", default="0", namespaces=NS)),
"evalue": float(hsp.findtext("n:evalue", default="0", namespaces=NS)),
"bit_score": float(hsp.findtext("n:bit-score", default="0", namespaces=NS)),
"score": int(hsp.findtext("n:score", default="0", namespaces=NS)),
"identity": int(hsp.findtext("n:identity", default="0", namespaces=NS)),
"align_len": int(hsp.findtext("n:align-len", default="0", namespaces=NS)),
"query_from": int(hsp.findtext("n:query-from", default="0", namespaces=NS)),
"query_to": int(hsp.findtext("n:query-to", default="0", namespaces=NS)),
"hit_from": int(hsp.findtext("n:hit-from", default="0", namespaces=NS)),
"hit_to": int(hsp.findtext("n:hit-to", default="0", namespaces=NS)),
# optional huge strings:
# "qseq": hsp.findtext("n:qseq", default="", namespaces=NS),
# "hseq": hsp.findtext("n:hseq", default="", namespaces=NS),
})
rec["hits"].append(hit_obj)
records.append(rec)
return records
# Example usage
data = parse_blastxml2("./MYYZBU9W016-Alignment.xml")
for rec in data:
print("Query:", rec["query_id"], rec["query_title"], "len=", rec["query_len"])
for hit in rec["hits"][:1]:
print("taxid=", hit["taxid"], "sciname=", hit["sciname"])
alignment in ChimeraX command line
import os
from chimerax.core.commands import run
target = r"E:/CLUSTERwork/PIEZO1_EVO/regionSearch/query4regions/blade1_1304.pdb"
inputFolder = r"E:/CLUSTERwork/PIEZO1_EVO/regionSearch/foldseekResults/chimeraX/bladePDB"
outputFolder = r"E:/CLUSTERwork/PIEZO1_EVO/regionSearch/foldseekResults/chimeraX/bladePDBaligned"
run(session, f'open "{target}"') # #1
for pdb_file in sorted(os.listdir(inputFolder)):
input_path = os.path.join(inputFolder, pdb_file)
output_path = os.path.join(outputFolder, pdb_file.replace(".pdb", "_aligned_CA.pdb"))
run(session, f'open "{input_path}"') # #2
run(session, 'mm #2@CA to #1@CA') # fit using only CA
run(session, 'select #2@CA') # keep only CA for saving
run(session, f'save "{output_path}" models #2 selectedOnly true')
run(session, 'close #2') # free memory
In chimeraX
runscript "E:/CLUSTERwork/PIEZO1_EVO/regionSearch/foldseekResults/chimeraX/chimeraX.py"
PyMol/ChimeraX
PyMol
- select residues based on residue number
select nterm, resi 42+74+43+56+39
- mutate one amino acid
cmd.wizard("mutagenesis")
cmd.fetch("target.pdb")
cmd.get_wizard().set_mode("LEU")
cmd.get_wizard().do_select("chain A and resid 22")
cmd.get_wizard().apply()
cmd.save("mutated_target.pdb")
- save as .pdb file.
File > Export Structure > Export Molecule > Save > change "save as type"
- select and color
select AFlow2, AF-A0A482RRZ8-F1-model_v6_aligned_CA and resi 57+147+148
color 0xFFDB13, AFlow2
- diagram
- Space-filling diagram:
- shows all atoms
- Ribbon/Cartoon diagram:
- shows the protein backbone and highlights the alpha helices.
- Surface diagram:
- shows areas that are accessible to water molecules.
- Space-filling diagram:
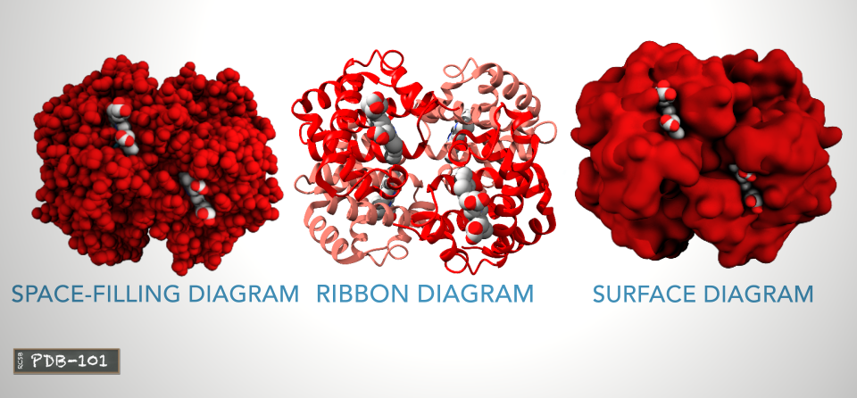
- select one atom
elect atom, ala and resi 4 and name CA
ChimeraX
color #1: 1305-1367 #00FE35- partial views
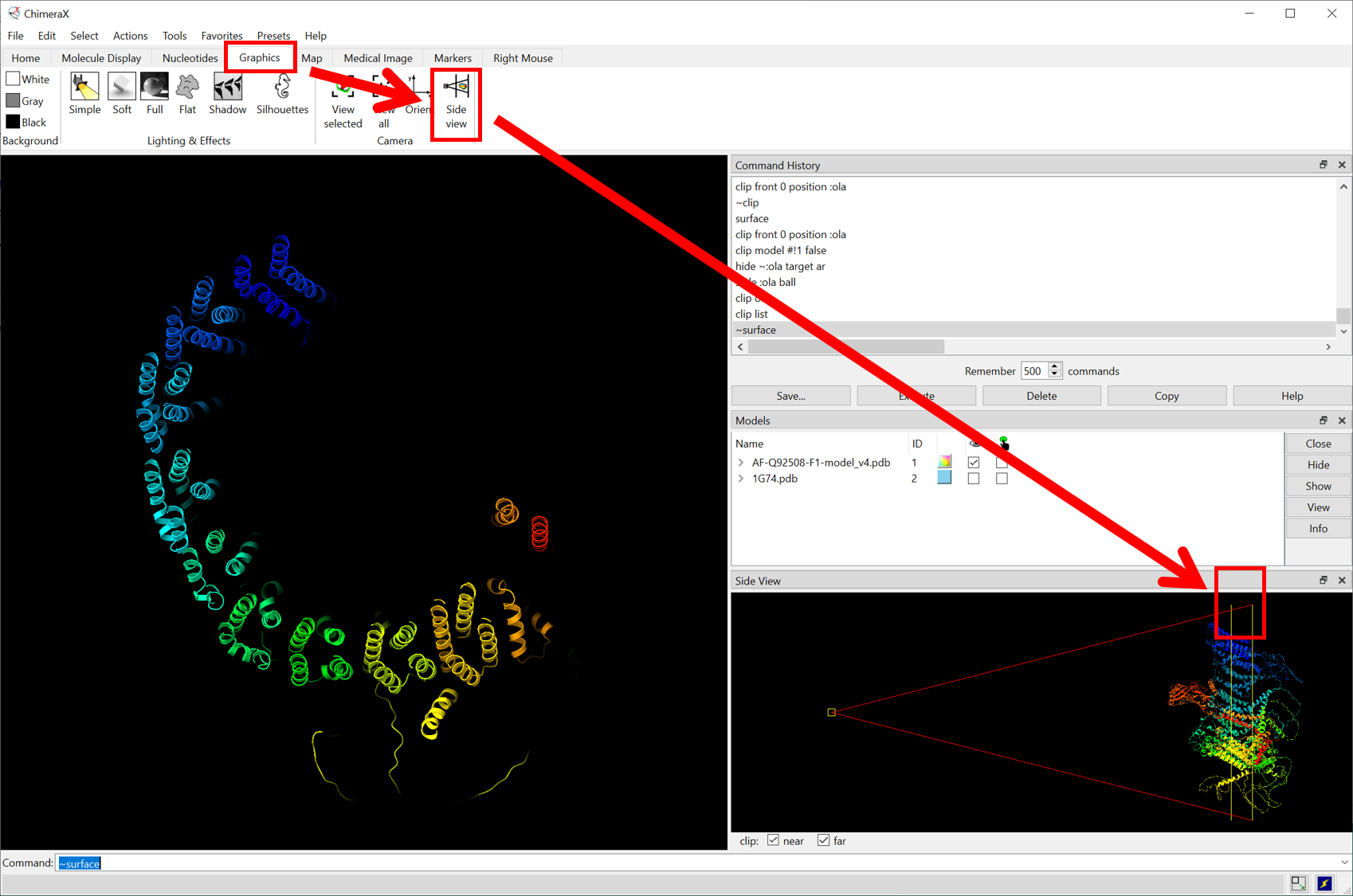
Molecular biology (Molecular Biology 5th (Fifth) Edition, Robert Weavers, amazon)
-
all living things are grouped into 3 domains:
1.1 bacteria 1.2 eukaryota 1.3 archaea -
Gene can exist in several different forms called allels (等位基因). Gene is the category, allel is the options under this category.
2.1 recessive 隐性 2.2 dominant 显性 2.3 heterozygote 杂合子 2.4 homozygote 纯合子
| Gene | Allele |
|---|---|
| A gene is a short sequence of DNA encoding for a certain trait | Alleles are different variants of a gene |
| Genes determine traits in an individual | Alleles are responsible for diverse features of a given trait |
| A gene can have many different alleles | An individual contains a pair of alleles for a particular gene, which may be homozygous or heterozygous, dominant or recessive |
| Examples: skin, hair, or eye colour | Examples: black, brown, blue, or green eye colour |
- Gene exist in a linear array on chromosomes.染色体
- Traits are governed by genes.
- If traits lie on the same chromosome, these traits can be inherited together.
- Homologous 同源 chromosomes are pairs (diploid cells) of chromosomes, one inherited from each parent.
- Meiosis happens during reproduction.
- In humans, we have 2 broad categories of chromosomes:
8.1 autosomes 常染色体, 22 different types.
8.2 sex chromosomes, X and Y

- The farther apart two genes lie on a chromosome, the more likely such recombination between them will be.
- In the transcription step, RNA polymerase makes a messenger RNA, which is a copty of the information in the gene. In the translation step, ribosomes read the mRNA and make the protein.
- DNA adn RNA are chain-like moleculars, and they are made of subunites called nucleotides.
- The nucleotides contain a base linked to 1'-position(one prime position) of a sugar (ribose in RNA or deoxyribose in DNA) and a phosphate group.
- The phosphate joins the sugars in a DNA or RNA chain through their 5'- and 3'-hydroxyl groups by phosphodiester bonds.
- adenin, thymin, guanine, cytosine.
- Certain viruses contain genes made of RNA instead of DNA.
- Proteins are polymers -- loong and chain-like moleculars.
- mRNA encodes a polypeptide. (mRNA codes fro a polypeptide)
- A linear chain of amino acid residues is called a polypeptide.
- Short polypeptides, containing less than 20–30 residues, are rarely considered to be proteins and are commonly called peptides.
- The amino acids in a polypeptide chain are linked by peptide bonds between amino (-NH2) and carboxyl group (-COOH).
- An individual amino acid in a chain is called a residue.
- A polypeptide chain ends with a free amino group, known as the N-terminus or amino terminus (positively charged), and a free carboxyl group, known as the C-terminus or carboxy terminus(negatively charged). polarity
- All proteins are made of 21 amino acids.
These amino acids can be divided into three groups based on their properties determnined by their side chains:
- Hydrophobic: carbon-rich side chains which don't interact well with water.
- Hydrophilic/Polar: iteract well with water.
- Charged: iteract with oppositely charded amino acids or other molecules.
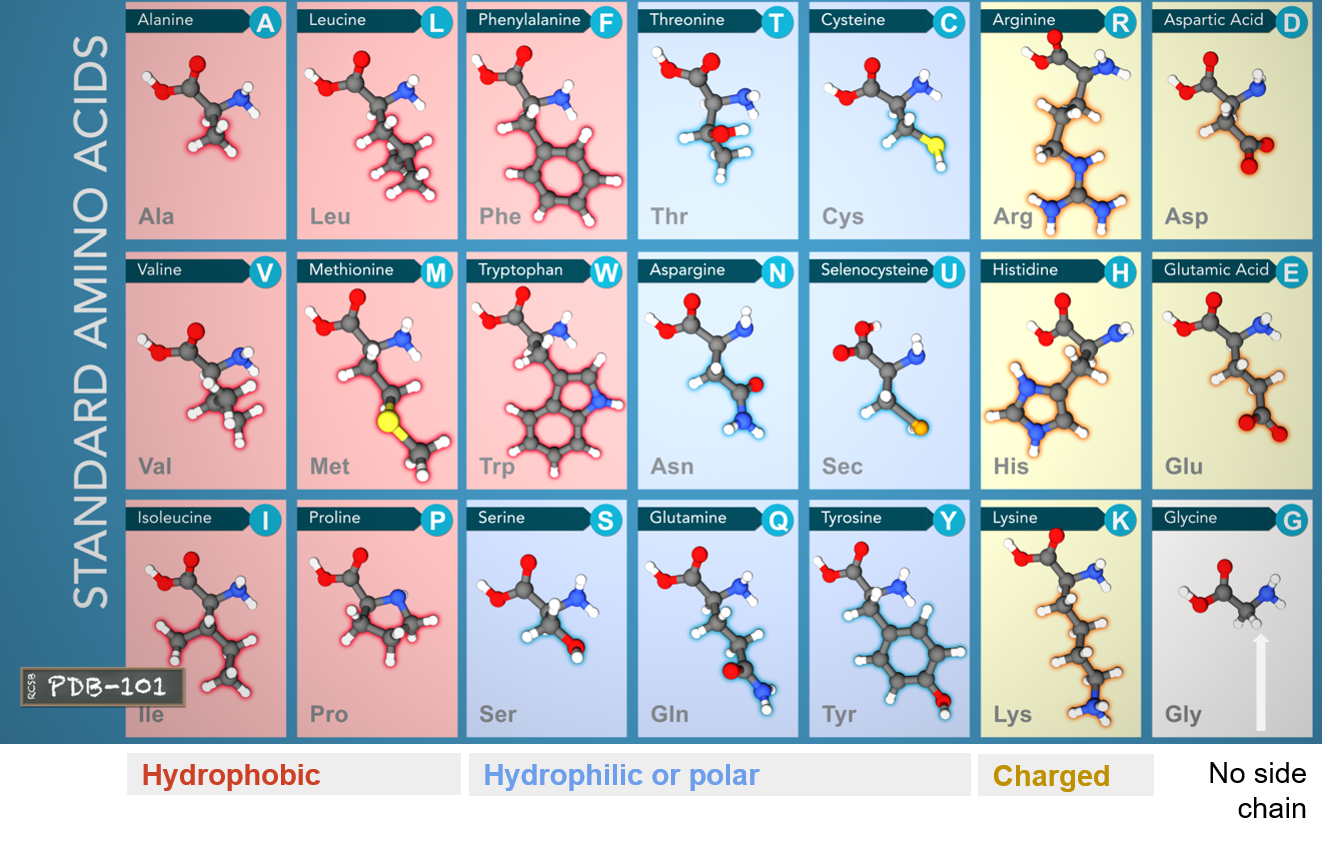
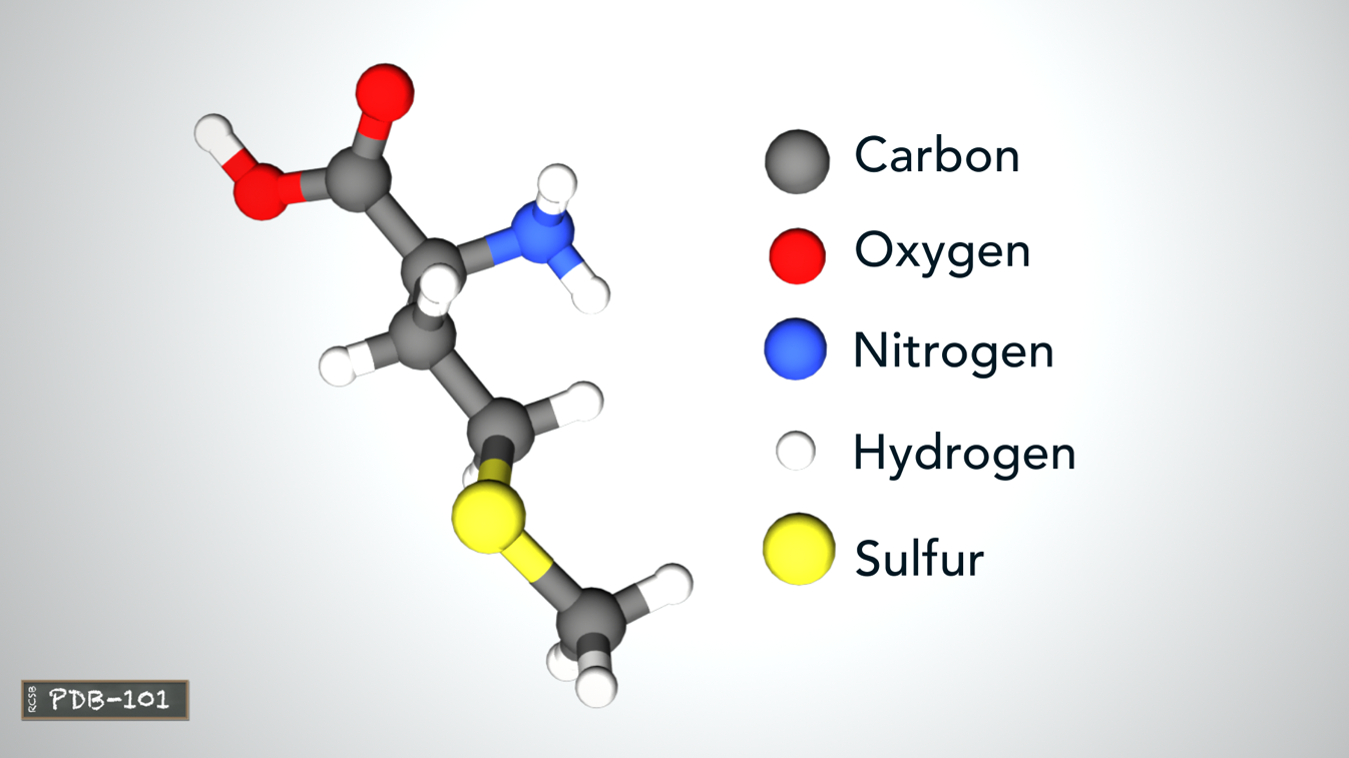
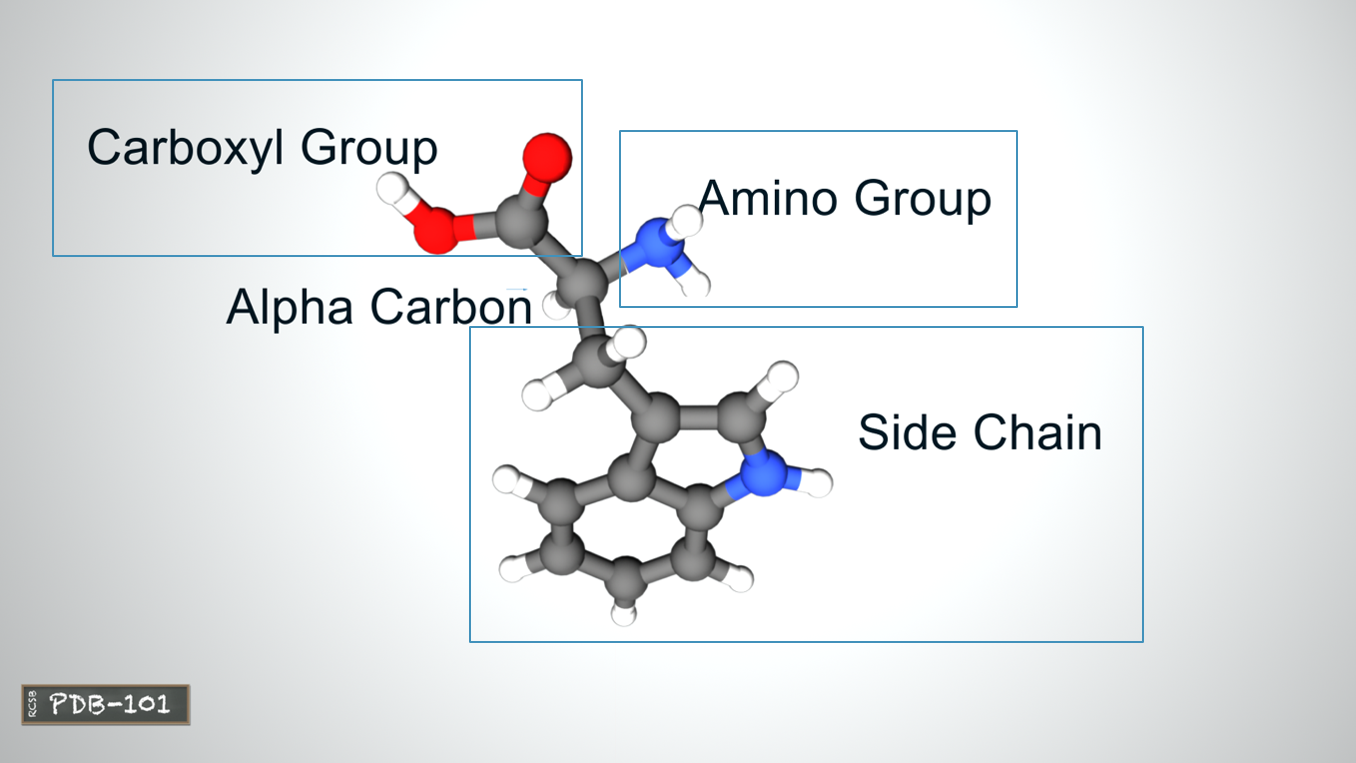
- A protein can be composed of one or more polypeptides.
- beta-strand vs beta-sheet
- Primary structure
Linear sequence of amino acids encoded by DNA. Amino acids are joined by peptide bonds, happending between -COOH and -NH2. A water H2O molecule is released each time a bond is formed. The linked series of carbon, nitrogen and oxygen atoms make up the protein backbone

- Secondary structure
Alpha helix and Beta sheet
- Tertiary structure
3D shape of the protein
- Quaternary structure
Two or more polypeptide chains come together to form one fucntional molecule wih several subunits.
reference
- Molecular biology (Molecular Biology 5th (Fifth) Edition, Robert Weavers, amazon)
- https://en.wikipedia.org/wiki/Protein
- https://pdb101.rcsb.org/learn/videos
A Deep learning Neural Network from the scratch via Numpy/Pandas
The basic complete deep learning neural network architecture includes:
- Forward propagation process with activation function (sigmoid and softmax) in the hidden layer and output laryer.
- Backward propagation process with derivative calculation and cross-entropy of each parameter (weight and bias).
- Update each parameter (weight and bias) simultaneously.
Task
- Goal: train a multiple-label classification neural network based on collected data.
- Method: handwritten the whole process.
- Expected output: the "difference" between the prediction and the reality can gradually decrease.
Dataset
3 samples, 2 features, 3 target labels.
| Sample | \(x_1\) | (x_2) | Target (label) |
|---|---|---|---|
| 1 | 0.04 | 0.42 | 0 |
| 2 | 1 | 0.54 | 1 |
| 3 | 0.5 | 0.65 | 2 |
Neural Network architecture







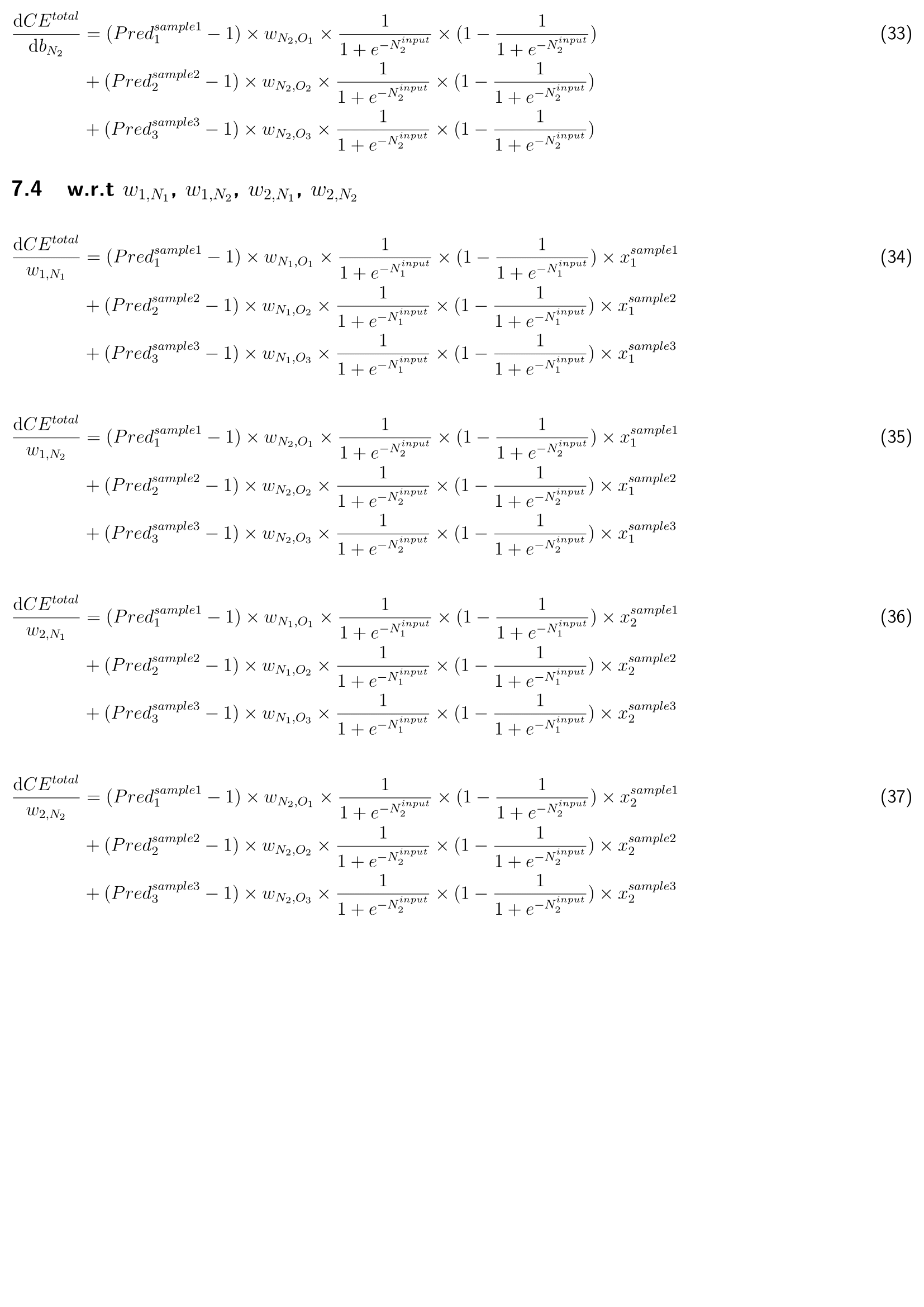
Paird t-Test
- Dependent sample \(t\)-test.
- It determineds whether the mean difference between two sets of observations is zero.
- One should measure each subject/entity twice, resulting in pairs of observations.
- Common application: Case-control studies, Repeat-measures desgins.
- Two competing hypotheses:
- Null hypothesis.
- assumes that the true mean difference between the paired samples is zero.
- all ovservanle differences are explained by random variation.
- Alternative hypothesis.
- assumes that the true mean difference between the paired samples is not equal to zero.
- explained:
- two-tailed: the true mean difference is not equal to zero.
- one-tailed:
- upper-tailed alternative hypothesis: the true mean difference is greater than zero.
- lower-tailed alternative hypothesis: the true mean difference is less than zero.
- Null hypothesis.
✅ 假设检验不是对你眼前这组数据下结论,而是试图推断产生这些数据的“背后机制”是什么。我们用数据去判断它更符合哪种假设:‘两组一样’还是‘有差异’。
A simple Bayesian inference example with Grid approximate, from scratch
why Bayesian?
Using probability methods to solve statistical problems.
For many problems in real life, we normally have an educated guess (prior), and this prior is helpful to our final guess/prediction. Through observing new data/evidence, we are upating our guess(prior), and the updated guess is posterior.
In the Bayesian inference workflow, we treat problems interatively, that is, we can get a new posterior through new observations, and then use it as the (new) prior.
Bayes' rule
Bayes' theorem begins with conditional probability.
because:
\[ Pr(A|B) = \frac{Pr(A\cap B)}{Pr(B)} \] \[ Pr(B|A) = \frac{Pr(B\cap A)}{Pr(A)} \]
then: \[ Pr(A|B)Pr(B) = Pr(A\cap B) \] \[ Pr(B|A)Pr(A) = Pr(B\cap A) \]
so: \[ Pr(A|B)Pr(B) = Pr(B|A)Pr(A) \]
Bayes' rule: \[ Pr(A|B) = \frac{Pr(B|A)Pr(A)}{Pr(B)} \]
Bayesian inference
The core of Bayesian Statistics is to update our understanding of unknown parameters through new observational data/evidence.
For example, we are curious to know a unknow (\theta), and we guess its prior is \(Pr(\theta)\). We have a sequence of observations (D). We hope \(D\) can help us better to understand \(Pr(\theta)\). It is that we are intersted in \(Pr(\theta|D)\). According to Bayes' rule, we know:
\[ Pr(\theta|D) = \frac{Pr(D|\theta)Pr(\theta)}{Pr(D)} \]
where:
- \(Pr(\theta)\): prior of \(\theta\), that is our guess of \(\theta\) without any observations \(D\).
- \(Pr(\theta|D)\): posterior of \(\theta\), that is our updated guess of \(\theta\) via any observations \(D\).
- \(Pr(D|\theta)\): likelihood, that is the probability of \(D\) under a certain \(\theta\).
- \(Pr(D)\): observations/evidence, that is the probability of \(D\) under all possible \(\theta\).
example
Statistical Rethinking by Richard McElreath
The example is from Statistical Rethinking by Richard McElreath. Here, I derive it from scratch and only using math.
1. QUESTION AND GOAL
There is a globe, and we want to know how much of the surface is covered in water.
You will toss the globe up in the air. When you catch it, you will record whether or not the surface under your right index finger is water \(W\) or land \(L\). Then you toss the globe up in the air again and repeat the procedure.
observations are: [ W \quad L\quad W\quad W\quad W\quad L\quad W\quad L\quad W ]
2. DATA INFORMATION
- The true proportion of water covering the globe is \(p\). (as \(\theta\) in the above work)
- A single toss of the globe has a probability \(p\) of producing a water (\(W\)) observation. It has a probability \(1-p\) of producing a land (\(L\)) observation. (There are only two events: \(W\) or \(L\))
- Each toss of the globe is independent of the others.
- The total number of observations is \(N=W+L\). In this case, \(N=9, W=6, L=3\).
3. MODELING
the assumption of the likelihood \(Pr(W,L|p)\):
\[ W \thicksim Binomail(N,p) \]
the binomial distribution is rather special for counting binary events.
About \(binomial\) distribution: \[ Pr(k,n,p)=Pr(X=k)=\frac{n!}{k!(n-k)!}p^k(1-p)^{(n-k)} \] The probability of getting exactly \(k\) successes in \(n\) independent \(Bernoulli\) trials (with the same rate \(p\)). In our case, the probability of seeing \(k=W\) in \(n=N=W+L\) observations is: \[ Pr(W,L,p)=Pr(X=W)=\frac{(W+L)!}{W!L!}p^W(1-p)^{L} \]
the prior of \(p\):
\[ Pr(p) \thicksim Uniform(0,1) \]
a flat distribution.
the posterior of \(p\) through observations \(W\) and \(L\):
\[ Pr(p|W,L) =\frac{Pr(W,L|p)Pr(p)}{Pr(W,L)} \]
4.GRID APPROXIMATE
the grid of \(p\) is \([0, 0.1, 0.3, 0.5, 1]\)
corresponding prior \(Pr(p)\) is \([1, 1, 1, 1, 1]\)
corresponding likelihood:
-
\(p_1=0\) \[ Pr(W,L|p_1)=\frac{(W+L)!}{W!L!}p_1^W(1-p_1)^{L}=0 \]
-
\(p_2=0.1\) \[ Pr(W,L|p_2)=\frac{(W+L)!}{W!L!}p_2^W(1-p_2)^{L}=\frac{9!}{6!3!}0.1^6\times 0.9^{3}=0.00006123600000000004 \]
-
\(p_3=0.3\) \[ Pr(W,L|p_3)=\frac{(W+L)!}{W!L!}p_3^W(1-p_3)^{L}=\frac{9!}{6!3!}0.1^6\times 0.9^{3}=0.02100394799999999 \]
-
\(p_4=0.5\) \[ Pr(W,L|p_4)=\frac{(W+L)!}{W!L!}p_4^W(1-p_4)^{L}=\frac{9!}{6!3!}0.1^6\times 0.9^{3}=0.1640625 \]
-
\(p_5=1\) \[ Pr(W,L|p_5)=\frac{(W+L)!}{W!L!}p_5^W(1-p_5)^{L}=\frac{9!}{6!3!}0.1^6\times 0.9^{3}=0 \]
\(Pr(W,L)\) (as \(Pr(D)\) in the above work):
\[
\begin{align*}
Pr(W,L) &= \displaystyle\sum_{i=1}^{5} Pr(W,L|p_i)\times Pr(p_i) \
&= 0\times 1 +0.00006123600000000004\times 1 + 0.02100394799999999\times 1 + 0.1640625\times 1 + 0\times 1\
&= 0.185127684
\end{align*}
\]
Pr(p|W,L)
-
\(p_1=0\) \[ Pr(p_1|W,L)=\frac{Pr(W,L|p_1)Pr(p_1)}{Pr(W,L)}=\frac{0\times 1}{ 0.185127684}=0 \]
-
\(p_2=0.1\) \[ Pr(p_2|W,L)=\frac{Pr(W,L|p_2)Pr(p_2)}{Pr(W,L)}=\frac{0.00006123600000000004\times 1}{ 0.185127684}=0.00033077710840913473 \]
-
\(p_3=0.3\) \[ Pr(p_3|W,L)=\frac{Pr(W,L|p_3)Pr(p_3)}{Pr(W,L)}=\frac{0.02100394799999999\times 1}{ 0.185127684}=0.11345654818433311 \]
-
\(p_4=0.5\) \[ Pr(p_4|W,L)=\frac{Pr(W,L|p_4)Pr(p_4)}{Pr(W,L)}=\frac{0.1640625\times 1}{ 0.185127684}=0.8862126747072577 \]
-
\(p_5=1\) \[ Pr(p_5|W,L)=\frac{Pr(W,L|p_5)Pr(p_5)}{Pr(W,L)}=\frac{0\times 1}{ 0.185127684}=0 \]
visualization
So..., I find I used these codes a lot.
git pull --rebase origin main
while IFS= read -r line; do
python ./../PDB_ana.py ${line} /../test_clean/
done < <(grep "" ./pdb.txt)
ls -l | wc -l
wget -i links.txt -o wget.log
tar -tzf file.tar.gz | wc -l # count files in tar.gz without unzip # it actually counts lines, and it will cause counting \n as a line
tar -zcvf myfolder.tar.gz myfolder
cp ./folder/*.txt ./newfolder
gunzip *.gz # for f in *.gz; do gunzip "$f"; done
cat *.fa > all_data.fa # concatenate multiple fasta files into one fasta file in command line
source ~/miniconda3/etc/profile.d/conda.sh
conda activate SE3nv
PDBdb_dir_has=()
for pdb_fp in $PDBdb_dir*pdb; do
pdb=$(basename $pdb_fp .pdb)
PDBdb_dir_has+=($pdb)
done
A=()
lines=`cat $AFDB_pLDDT_70_log`
for line in $lines; do
if [[ $line == *".pdb" ]]; then
entry=$(echo "$line" | cut -d "." -f 1)
A+=($entry)
fi
done
How to discard changes in VScode?
Navigate to reporsitory (has .github folder) with changes. Then, git restore <file>
Activate conda environment in bash script
source ~/miniconda3/etc/profile.d/conda.sh
conda activate ENV
SBATCH Array jobs
#SBATCH -J array-job
#SBATCH -a 1-3
#SBATCH -o array-job.%A.%a.log
id_list="./id_list.txt"
id=`head -n $SLURM_ARRAY_TASK_ID $id_list | tail -n 1`
python3 script.py -in $id
srun gpu
srun -N 1 --ntasks-per-node 2 -p v100 -q gpu --gres=gpu:1 --mem=50G --pty /bin/bash
run scripts
sbatch test.slm
check sequence process
squeue -u username
cancel sequence
scancel jobID
copy files into another folder
cp ./*.pdb ../newfoler
count files in a directory in command line
cd directory
ls -l | wc -l
# get .gz files
wget -r -np -e robots=off -A 'pep.all.fa.gz' https://ftp.ensembl.org/pub/current_fasta/
stat_gpu # check available partition
srun -q gpu -p v100-af --mem=50G --gres=gpu:1 --pty /bin/bash # access one gpu
tar xvzf file.tar.gz # unzip
module load igv # access specific place
ls -1 | wc -l # count files in a directory in linux

Overleaf+Latx+Zotero
https://medium.com/@nooraaz/use-zotero-in-overleaf-without-premium-251fcfd7f369
https://retorque.re/zotero-better-bibtex/
% paragraph spacing
\setlength{\parskip}{1em} % Space between paragraphs
\setlength{\parindent}{0em} % Indentation at the beginning of each paragraph
Assign files into different folders
Assuming I have 230 files in db_folder, and I want to copy these 230 files into different folders (folder0, folder1 and so on) with at least 50 files in each folder.
files_list contains names of these 230 files, like files_list=["file1", "file2", ..., "file230"]
import shutil
chunks = [files_list[x:x+50] for x in range(0, len(files_list), 50)]
# len(chunks) = 5
for i in range(53):
chunk = chunks[i]
newpath = "db_folder/folder" + str(i)
if not os.path.exists(newpath):
os.makedirs(newpath)
for j in chunk:
src = "db_folder/" + j
dst = "db_folder/folder" + str(i) + "/" + j
shutil.copyfile(src, dst)
Copy files to a new folder
import shutil
for i in files_list:
src = "old_folder/" + i
dst = "new_folder/" + i
shutil.copyfile(src, dst)
Remove legend in Plotly
fig.update_layout(showlegend=False)
unzip compressed files into a new directory
gz_file = os.path.join(root, file)
out_file = "new_path/" + file
with gzip.open(gz_file,"rb") as f_in, open(out_file,"wb") as f_out:
shutil.copyfileobj(f_in, f_out)
copy and move
import shutil
shutil.copyfile("old_path/file.txt", "new_path/file.txt")
Visualization
Plotly: Drawing a plane perpendicular to a given line
- Explanation
- Example
When I was working on my binder design task, I need to select suitable hotspot residues that can lead to design better binders. In addition to hydrophobic amino acids, I also want to focus on the amino acids whose positions are close to the outside edge, and far away from the center of the protein. Therefore, I expect a plane that can approximately cut the protein layer by layer with my expected distance to the outside edge.
By calculating the centers of two parts of my protein, I can easily determine one line. However, it took me much time to find the plane that can be perpendicular to this line.
Finally, I find the solution!
Planes perpendicular to the given line
Explanation
Assuming that we want to make a plane whose vertical distance to \(p_1\) is \(d_{01}\). We can determine the point \(p_0) by
from shapely.geometry import LineString
line = LineString([p1, p2])
p0_tmp = line.interpolate(d01)
p0 = np.array([p0_tmp.x, p0_tmp.y, p0_tmp.z])
5 important points who determine the plane
Finding \(p_3\), \(p_4\), \(p_5\) and \(p_6\) took me lots of time. I finally find that they can be found by vector calculation. For example, we want the distance between \(p_0\) and \(p_3\) or \(p_4\) or \(p_5\) or \(p_6\) is \(Radius = 3\).
P1 = p0
P2 = p2
Radius = 3
V3 = P1 - P2
V3 = V3 / np.linalg.norm(V3)
e = np.array([0,0,0])
e[np.argmin(np.abs(V3))] = 1
V1 = np.cross(e, V3)
V1 = V1 / np.linalg.norm(V3)
V2 = np.cross(V3, V1)
# p3 90 degree
s3 = np.pi/2
p3 = P1 + Radius*( np.cos(s3)*V1 + np.sin(s3)*V2 )
# p4 180 degree
s4 = np.pi
p4 = P1 + Radius*( np.cos(s4)*V1 + np.sin(s4)*V2 )
# p5 270 degree
s5 = np.pi*1.5
p5 = P1 + Radius*( np.cos(s5)*V1 + np.sin(s5)*V2 )
# p6 0 degree
s6 = 0
p6 = P1 + Radius*( np.cos(s6)*V1 + np.sin(s6)*V2 )
Becasue three points determine one plane. We can finally make the plane by
plane1_x, plane1_y, plane1_z = np.array([p6, p3, p5]).T
fig.add_trace(go.Mesh3d(x=plane1_x, y=plane1_y, z=plane1_z, color="lightgreen", name = "", hoverinfo="skip", opacity=0.50))
plane2_x, plane2_y, plane2_z = np.array([p4, p3, p5]).T
fig.add_trace(go.Mesh3d(x=plane2_x, y=plane2_y, z=plane2_z, color="lightgreen", name = "", hoverinfo="skip", opacity=0.50))
Example
importlibraries
import numpy as np
from random import randrange
import random
import pandas as pd
from shapely.geometry import LineString
import plotly.express as px
import plotly.graph_objects as go
- There are two points \(p_1\) and \(p_2\), and I want a plane who is perpendicular to the line \( (p_1,p_2) \) at the point \(p_0\). The vertical distance between \(p_1\) to this plane is the distance between \(p_1\) and \(p_0\), \(d_{01}=4\).
random.seed(25)
p1 = np.array([randrange(1,10) for i in range(3)])
p2 = np.array([randrange(3,15) for i in range(3)])
d01 = 5
- Half the length of the diagonal of the square plane is \(r=5\)
def drawing(p1, p2, d01, r):
# p0 position
line = LineString([p1, p2])
p0_tmp = line.interpolate(d01)
p0 = np.array([p0_tmp.x, p0_tmp.y, p0_tmp.z])
# preparation
P1 = p0
P2 = p2
Radius = r
V3 = P1 - P2
V3 = V3 / np.linalg.norm(V3)
e = np.array([0,0,0])
e[np.argmin(np.abs(V3))] = 1
V1 = np.cross(e, V3)
V1 = V1 / np.linalg.norm(V3)
V2 = np.cross(V3, V1)
# p3 90 degree
s3 = np.pi/2
p3 = P1 + Radius*( np.cos(s3)*V1 + np.sin(s3)*V2 )
# p4 180 degree
s4 = np.pi
p4 = P1 + Radius*( np.cos(s4)*V1 + np.sin(s4)*V2 )
# p5 270 degree
s5 = np.pi*1.5
p5 = P1 + Radius*( np.cos(s5)*V1 + np.sin(s5)*V2 )
# p6 0 degree
s6 = 0
p6 = P1 + Radius*( np.cos(s6)*V1 + np.sin(s6)*V2 )
# draw
## line
fig = go.Figure(data =[go.Scatter3d(x=[p1[0],p2[0]], y=[p1[1], p2[1]],z=[p1[2], p2[2]],
mode = "markers+lines+text",
marker=dict(color="gray", size=15, symbol="cross"),
line=dict(color="gray", width=5),
text=["p1", "p2"],
textfont=dict(size=20))])
## plane
plane1_x, plane1_y, plane1_z = np.array([p6, p3, p5]).T
fig.add_trace(go.Mesh3d(x=plane1_x, y=plane1_y, z=plane1_z, color="lightgreen", name = "", hoverinfo="skip", opacity=0.50))
plane2_x, plane2_y, plane2_z = np.array([p4, p3, p5]).T
fig.add_trace(go.Mesh3d(x=plane2_x, y=plane2_y, z=plane2_z, color="lightgreen", name = "", hoverinfo="skip", opacity=0.50))
fig.update_layout(
width=1100, height=1100,
scene=dict(xaxis_title='x coord',yaxis_title='y coord',zaxis_title='z coord'),showlegend=False)
#fig.show()
# ddataframe
df = pd.DataFrame(columns=["name", "x", "y", "z", "c"])
df["name"] = ["p0", "p1", "p2", "p3", "p4", "p5", "p6"]
df["x"] = [p0[0], p1[0], p2[0], p3[0], p4[0], p5[0], p6[0]]
df["y"] = [p0[1], p1[1], p2[1], p3[1], p4[1], p5[1], p6[1]]
df["z"] = [p0[2], p1[2], p2[2], p3[2], p4[2], p5[2], p6[2]]
df["c"] = ["orange", "gray", "gray", "red", "blue", "green", "deeppink"]
fig.add_trace(
go.Scatter3d(x = [p0[0]],
y = [p0[1]],
z = [p0[2]],
mode ='markers+text',
marker=dict(color="gray", size=15, symbol="cross"),
text=["p0"],
textfont=dict(size=20)))
fig.show()
return df
df = drawing(p1=p1, p2=p2, d01=d01, r=5)
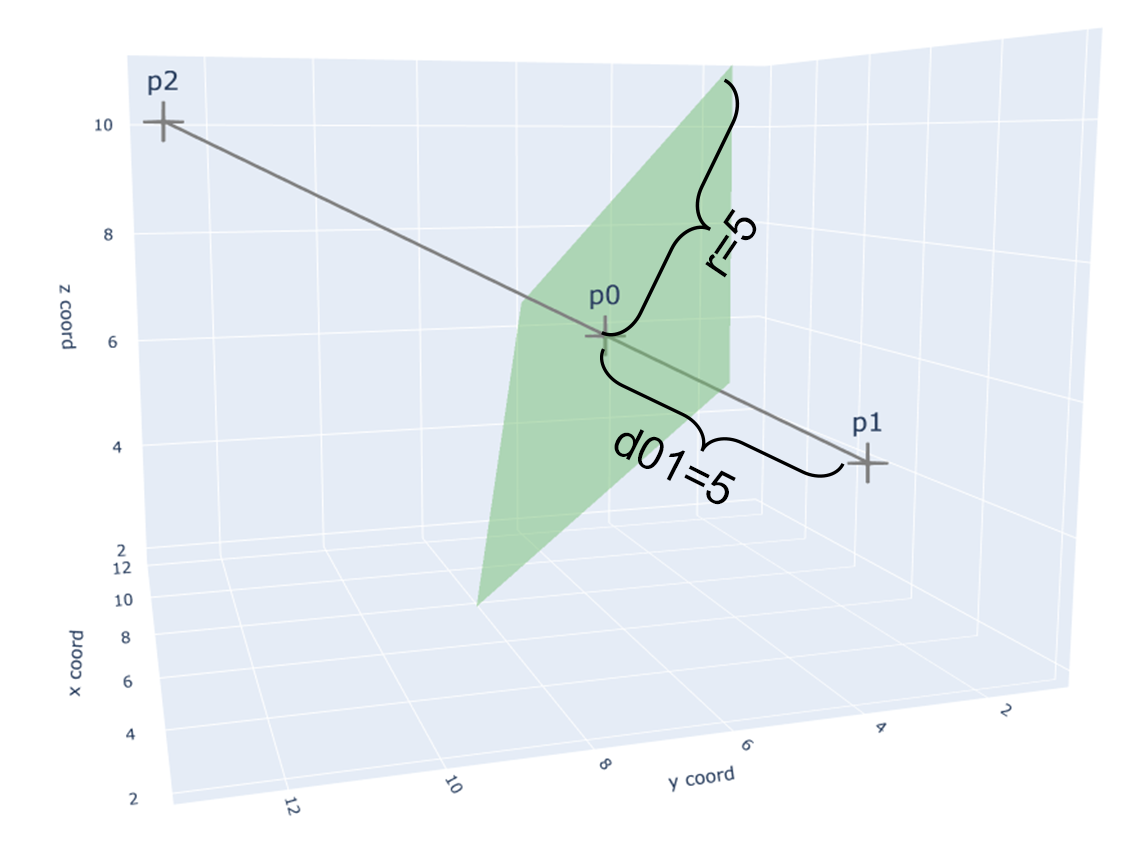
A square plane perpendicular to a line
- Many plans
random.seed(25)
p1 = np.array([randrange(1,10) for i in range(3)])
p2 = np.array([randrange(3,17) for i in range(3)])
d01_list = [1,2,3,4,5,6]
r_list = [7,6,5,4,3,2]
color = ["lightgreen", "lightpink", "lightskyblue", "lightgray", "lightcoral", "lightgoldenrodyellow"]
## line
fig_base = go.Figure(data =[go.Scatter3d(x=[p1[0],p2[0]], y=[p1[1], p2[1]],z=[p1[2], p2[2]],
mode = "markers+lines",
marker=dict(color="gray", size=15, symbol="cross"),
line=dict(color="gray", width=5))])
fig_base.update_layout(
width=1100, height=1100,
scene=dict(xaxis_title='x coord',yaxis_title='y coord',zaxis_title='z coord'),showlegend=False)
for d01, r, c in zip(d01_list, r_list, color):
# p0 position
line = LineString([p1, p2])
p0_tmp = line.interpolate(d01)
p0 = np.array([p0_tmp.x, p0_tmp.y, p0_tmp.z])
# preparation
P1 = p0
P2 = p2
Radius = r
V3 = P1 - P2
V3 = V3 / np.linalg.norm(V3)
e = np.array([0,0,0])
e[np.argmin(np.abs(V3))] = 1
V1 = np.cross(e, V3)
V1 = V1 / np.linalg.norm(V3)
V2 = np.cross(V3, V1)
# p3 90 degree
s3 = np.pi/2
p3 = P1 + Radius*( np.cos(s3)*V1 + np.sin(s3)*V2 )
# p4 180 degree
s4 = np.pi
p4 = P1 + Radius*( np.cos(s4)*V1 + np.sin(s4)*V2 )
# p5 270 degree
s5 = np.pi*1.5
p5 = P1 + Radius*( np.cos(s5)*V1 + np.sin(s5)*V2 )
# p6 0 degree
s6 = 0
p6 = P1 + Radius*( np.cos(s6)*V1 + np.sin(s6)*V2 )
# draw
## line
fig = go.Figure(data =[go.Scatter3d(x=[p1[0],p2[0]], y=[p1[1], p2[1]],z=[p1[2], p2[2]],
mode = "markers+lines",
marker=dict(color="gray", size=15, symbol="cross"),
line=dict(color="gray", width=5))])
## plane
plane1_x, plane1_y, plane1_z = np.array([p6, p3, p5]).T
fig_base.add_trace(go.Mesh3d(x=plane1_x, y=plane1_y, z=plane1_z, color=c, name = "", hoverinfo="skip", opacity=0.50))
plane2_x, plane2_y, plane2_z = np.array([p4, p3, p5]).T
fig_base.add_trace(go.Mesh3d(x=plane2_x, y=plane2_y, z=plane2_z, color=c, name = "", hoverinfo="skip", opacity=0.50))
fig_base.show()
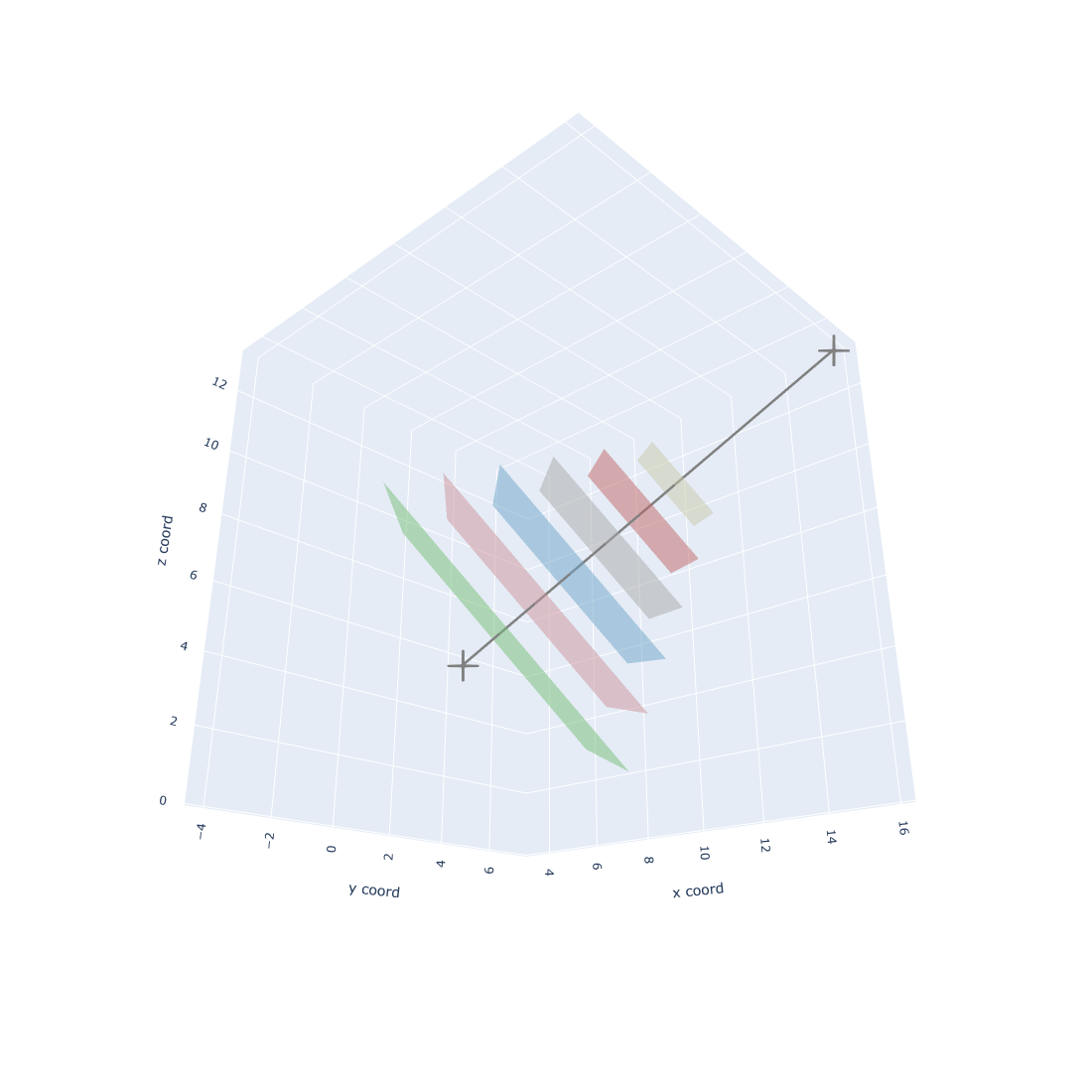
Planes perpendicular to the line
Plotly: Copy&Paste Continuous and Discrete colors
Discrete color names
 |
aliceblue |  |
antiquewhite |
 |
aqua |  |
aquamarine |
 |
azure |  |
beige |
 |
bisque |  |
black |
 |
blanchedalmond |  |
blue |
 |
blueviolet |  |
brown |
 |
burlywood |  |
cadetblue |
 |
chartreuse |  |
chocolate |
 |
coral |  |
cornflowerblue |
 |
cornsilk |  |
crimson |
 |
cyan |  |
darkblue |
 |
darkcyan |  |
darkgoldenrod |
 |
darkgray |  |
darkgrey |
 |
darkgreen |  |
darkkhaki |
 |
darkmagenta |  |
darkolivegreen |
 |
darkorange |  |
darkorchid |
 |
darkred |  |
darksalmon |
 |
darkseagreen |  |
darkslateblue |
 |
darkslategray |  |
darkslategrey |
 |
darkturquoise |  |
darkviolet |
 |
deeppink |  |
deepskyblue |
 |
dimgray |  |
dimgrey |
 |
dodgerblue |  |
firebrick |
 |
floralwhite |  |
forestgreen |
 |
fuchsia |  |
gainsboro |
 |
ghostwhite |  |
gold |
 |
goldenrod |  |
gray |
 |
grey |  |
green |
 |
greenyellow |  |
honeydew |
 |
hotpink |  |
indianred |
 |
indigo |  |
ivory |
 |
khaki |  |
lavender |
 |
lavenderblush |  |
lawngreen |
 |
lemonchiffon |  |
lightblue |
 |
lightcoral |  |
lightcyan |
 |
lightgoldenrodyellow |  |
lightgray |
 |
lightgrey |  |
lightgreen |
 |
lightpink |  |
lightsalmon |
 |
lightseagreen |  |
lightskyblue |
 |
lightslategray |  |
lightslategrey |
 |
lightsteelblue |  |
lightyellow |
 |
lime |  |
limegreen |
 |
linen |  |
magenta |
 |
maroon |  |
mediumaquamarine |
 |
mediumblue |  |
mediumorchid |
 |
mediumpurple |  |
mediumseagreen |
 |
mediumslateblue |  |
mediumspringgreen |
 |
mediumturquoise |  |
mediumvioletred |
 |
midnightblue |  |
mintcream |
 |
mistyrose |  |
moccasin |
 |
navajowhite |  |
navy |
 |
oldlace |  |
olive |
 |
olivedrab |  |
orange |
 |
orangered |  |
orchid |
 |
palegoldenrod |  |
palegreen |
 |
paleturquoise |  |
palevioletred |
 |
papayawhip |  |
peachpuff |
 |
peru |  |
pink |
 |
plum |  |
powderblue |
 |
purple |  |
red |
 |
rosybrown |  |
royalblue |
 |
saddlebrown |  |
salmon |
 |
sandybrown |  |
seagreen |
 |
seashell |  |
sienna |
 |
silver |  |
skyblue |
 |
slateblue |  |
slategray |
 |
slategrey |  |
snow |
 |
springgreen |  |
steelblue |
 |
tan |  |
teal |
 |
thistle |  |
tomato |
 |
turquoise |  |
violet |
 |
wheat |  |
white |
 |
whitesmoke |  |
yellow |
 |
yellowgreen |
Continuous color names
 |
aggrnyl |
 |
agsunset |
 |
blackbody |
 |
bluered |
 |
blues |
 |
blugrn |
 |
bluyl |
 |
brwnyl |
 |
bugn |
 |
bupu |
 |
burg |
 |
burgyl |
 |
cividis |
 |
darkmint |
 |
electric |
 |
emrld |
 |
gnbu |
 |
greens |
 |
greys |
 |
hot |
 |
inferno |
 |
jet |
 |
magenta |
 |
magma |
 |
mint |
 |
orrd |
 |
oranges |
 |
oryel |
 |
peach |
 |
pinkyl |
 |
plasma |
 |
plotly3 |
 |
pubu |
 |
pubugn |
 |
purd |
 |
purp |
 |
purples |
 |
purpor |
 |
rainbow |
 |
rdbu |
 |
rdpu |
 |
redor |
 |
reds |
 |
sunset |
 |
sunsetdark |
 |
teal |
 |
tealgrn |
 |
turbo |
 |
viridis |
 |
ylgn |
 |
ylgnbu |
 |
ylorbr |
 |
ylorrd |
 |
algae |
 |
amp |
 |
deep |
 |
dense |
 |
gray |
 |
haline |
 |
ice |
 |
matter |
 |
solar |
 |
speed |
 |
tempo |
 |
thermal |
 |
turbid |
 |
armyrose |
 |
brbg |
 |
earth |
 |
fall |
 |
geyser |
 |
prgn |
 |
piyg |
 |
picnic |
 |
portland |
 |
puor |
 |
rdgy |
 |
rdylbu |
 |
rdylgn |
 |
spectral |
 |
tealrose |
 |
temps |
 |
tropic |
 |
balance |
 |
curl |
 |
delta |
 |
oxy |
 |
edge |
 |
hsv |
 |
icefire |
 |
phase |
 |
twilight |
 |
mrybm |
 |
mygbm |
Code
More
https://plotly.com/python/builtin-colorscales/
mdbook usage
<center>
<figure>
<img src="./img/uniprot.png" alt=" " width="600">
<figcaption>Fig.1 the Entry from UniProt</figcaption>
</figure>
</center>
<iframe
src="https://huggingface.co/datasets/tattabio/OMG_prot50/embed/viewer/default/train"
frameborder="0"
width="90%"
height="560px"
></iframe>
In Windows10, build personal website via Hugo-book and Github Actions
This is about how to use Hugo to create the local website, and then deploy it on GitHub via Github Action.
Prerequisites
- Git
- Windows 10
- VScode
- Github account
Install Hugo
- In Windows10, right click Windows Powershell and click run as administrator.
- Refer to How to install chocolatey in Windows to install chocolatey.
- Refer to Install Hugo on Windows to install Hugo by
choco install hugo-extended
- Make sure Hugo works well by
hugo version
and we will get the following
hugo v0.123.7-312735366b20d64bd61bff8627f593749f86c964+extended windows/amd64 BuildDate=2024-03-01T16:16:06Z VendorInfo=gohugoio
Create the website using Hugo themes
For example, I want to locally work on my website in C:\myweb folder. Still in Windows Powershell,
- Navigate back to
C:driver, and create a new folder by typingmkdir myweb. - Create the website by
hugo new site Blogs
cd Blogsgit init- Refer to Hugo Book Theme to use hugo-book theme by
git submodule add https://github.com/alex-shpak/hugo-book themes/hugo-book
- Till now, if we run
hugo server, we will get a plain website locally.
In VS code, open Blogs folder.
- In
C:/myweb/Blogs/themes/hugo-book/exampleSite/content.en/, copydocsfolder,postsfolder and_index.mdfile toC:/myweb/Blogs/content/.
Why I don't copy the
menufolder?
Because I foundmenufolder doesn't work when I modified the website as I needed. We can customize the order of menus in leftbar by adding weight in thename.mdfile. For example, in eachname.mdfile, the parameterweightdetermine the order of menues.--- weight: 1 title: One name.md file ---
- Run
hugo serveragain, we will see a more well-done website.
Deploy on Github by Action
- Create a public Github repository.
For example, if this repository's name is Blogs, your website link will be like this https://huilin-li.github.io/Blogs/
- Still in Windows Powershell, push your local work to this repository by
cd C:/myweb/Blogs
git add .
git commit -m "update"
git branch -M main
git remote add origin https://github.com/Huilin-Li/Blogs.git
git push -u origin main
- Visit your GitHub repository. From the main menu choose Settings > Pages. In the center of your screen you will see this:
{{< figure src="../images/deploy1.png" width="400" alt="deploy1" >}}
- Change the Source to
GitHub Actions. - Click
Configureas the highlight in this picture:
{{< figure src="../images/deploy2.png" width="400" alt="deploy2" >}}
- Go to Host on GitHub Pages, copy the
yamlfile instep 6toBlogs/.github/workflows/hugo.yaml, and commit changes. - As Step8, Step9, Step10 in Host on GitHub Pages, the deloyment is done. 🎉
Update website
Add more contents in C:/myweb/Blogs/content/ folder, and then git push to Github repository. Github Action will automaticaly update new contents in https://huilin-li.github.io/Blogs/.
References
- How to install chocolatey in Windows
- Hugo with Git Hub Pages on Windows
- Using GitHub Pages with Actions to deploy Hugo sites in seconds - Tommy Byrd // HugoConf 2022
- Hugo Book Theme
- Host on GitHub Pages
- 【Hugo】hugo-book主题使用
Hugo
SUMMARY
- use themes as your own repositories
- insert and resize images
- center images
- add emoji
- add travel map
- Why my local website doesn't update after I update my markdown files?
- link pages and tiles
- Add Giscus comment in Hugo-book theme
Remove git in themes, and use themes as your own repositories.
git rm --cached hugo-book -f
So that, we could modify, git add, and git push the theme repository as our own.
How to insert images and resize images?
The file tree is like
├─content
│ │ _index.md
│ └─posts
│ │ hugo.md
│ │ _index.md
│ └─images
│ Nice.png
In hugo.md file, we add Nice.png and scale it by
{{< figure src="../images/Nice.png" width="400" alt=" ">}}
Why we need to add ../ in front of images folder, altough hugo.md file and images folder are at the same level?
Because Hugo system sees hugo.md as a foler too. However, Hugo system sees _index.md as a file! Therefore, If we want to insert iamges in _index.md file, the command will be like:
{{< figure src="../images/Nice.png" width="400" alt=" " >}}
Center the image?
<center>{{<figure src="../images/Nice.png" width="400" alt=" ">}}</center>
At the same time, in hugo.toml, add
[markup]
[markup.goldmark]
[markup.goldmark.renderer]
unsafe = true
How to add emoji?
Directly copy emoji icon 👋 and paste to .md file, instead of using :wave:.
Emoji Copy&Paste: https://emojidb.org/number-emojis
How to add a travel map in Hugo?
Refer to this article
NOTE:
In {{< openstreetmap mapName="<your map name>" >}}, the <your map name> is not the name you give to your map, but the name in the website link of your map. For example, I created a map whose name is Travel, I need to use travel_1036974 in the hugo shortcode.
{{< figure src="../images/map.jpeg" width="400" alt="umap">}}
Why my local website doesn't update after I update my markdown files?
It works well if I Press Ctrl+C to stop the local host, and hugo server again, I can see the updated website in http://localhost:1313/.
How to link pages and tiles?
I am in now.md file now, and there is another page.md which has #Title I want to link. I want to link to page.md and link to #Title I want to link.
In page.md file, add an anchor:
# Title I want to link {#anchor}
In now.md file, add commands:
[text]({{< ref "./pages.md" >}}) # link to one page
[text]({{< ref "./pages.md#anchor" >}}) # link to the title
Add Giscus comment in Hugo-book theme
- Create a new public github repository, e.g.
site-comment. - Enable the discussions feature.
- Install the giscus app
For example,
- Configure giscus codeblock. Go to Giscus App, and get the anable Giscus.
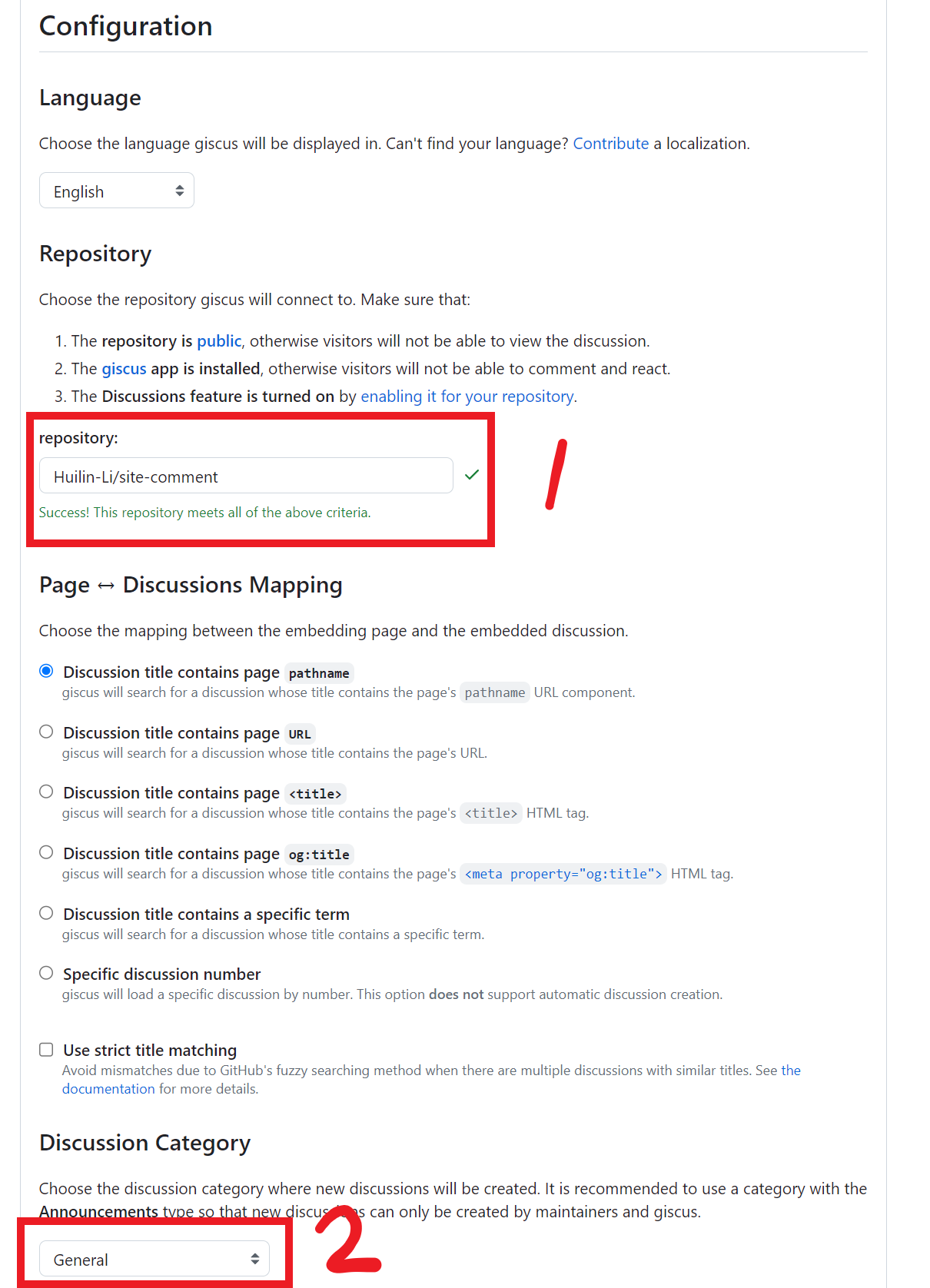
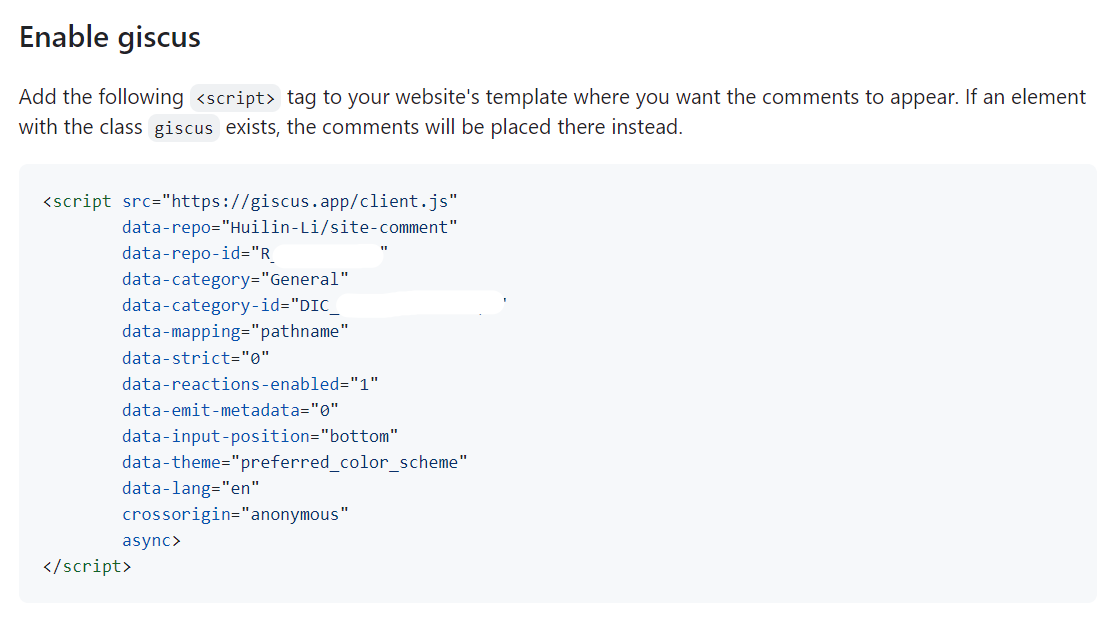
- Add the
scripttothemes\hugo-book\layouts\partials\docs\comments.html, like this
References
- hugo book demo
- 【Hugo】hugo-book主题使用
- hugo博客 文内插入图片
- How to render markdown url with .md to correct link
- Linking pages in Hugo/markdown
- Adding comments system to a Hugo site using Giscus
- https://genomicsclass.github.io/book/
- https://leanpub.com/dataanalysisforthelifesciences/
- https://www.statlearning.com/
- https://www.huber.embl.de/msmb/
Contact
- Outlook: LHLLIHUILIN@outlook.com
- UniEmail: huilinli@bmb.sdu.dk
- Gmail: lihuilin023@gmail.com