Design binders via RFdiffusion and ProteinMPNN-FastRelax
In my use case, firstly, I tried to de novo design binders. However, I found that designed binders are limited to proteins with only one helix. Then, I changed to design binders based on scaffolds. The design process and scripts I used a lot are as follows.
Three needed designing environments
Thanks a lot to the great work, RFdiffusion and ProteinMPNN-FastRelax, as well as AlphaFold2, who give us great ways to design proteins.
- RFdiffuion for desigining protein backbones, under
SE3nv environment. - ProteinMPNN-FastRelax for desiging protein sequences, under
dl_binder_design environment. - AlphaFold2 for evaluating designed proteins, under
af2_binder_design environment.
We will design binders under these three environments: SE3nv environment, dl_binder_design environment, af2_binder_design environment.
Install these three environments
Make sure you have high performance computating envrionment with CUDA, and you can access GPU and internet. Their installations can automatically install dependencied packages that fit the GPU environment which you have to use for the heavy computation.Quickly go through the whole process
Let us say that we want to design binders for target.pdb, and our current directories tree is
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── target
│ │ └── target.pdb
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
1️⃣ get secondary structure and block adjacency information script get_adj_secstruct.slm
We will provide these two .pt files (target_adj.pt and target_ss.pt) to the scaffold-based binder design model.
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -o %j.out
source ~/miniconda3/etc/profile.d/conda.sh
conda activate SE3nv
cd /(root)/mydesigns
python3 ../RFdiffusion/helper_scripts/make_secstruc_adj.py --input_pdb ./target/target.pdb --out_dir ./target_adj_secstruct
Then, our directories tree is updated as:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
2️⃣ backbones design script bb.slm
I use array-job to parallelly execute different ppi.hotspot_res arguments simultaneously. For example, I have 10 ppi.hotspot_res arguments to test, so my paraID.txt is like:
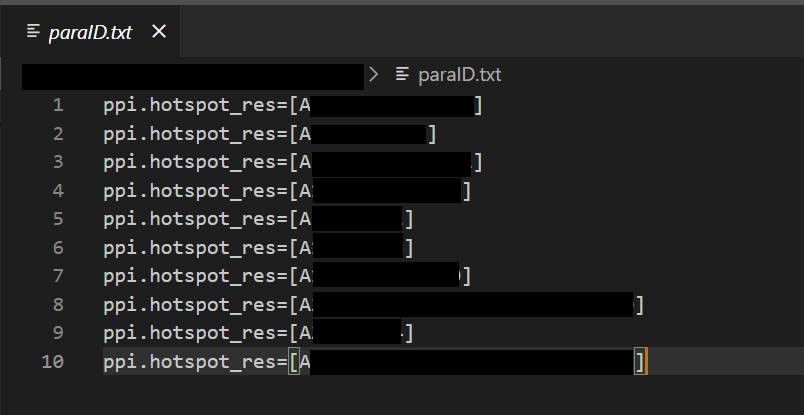
and I setup 10 jobs (#SBATCH -a 1-10) for each ppi.hotspot_res. Each ppi.hotspot_res will design inference.num_designs=10000 binder backbones.
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -J array-job
#SBATCH -a 1-10
#SBATCH -o backbone_10000.%A.%a.log
id_list="./paraID.txt"
id=`head -n $SLURM_ARRAY_TASK_ID $id_list | tail -n 1`
source ~/miniconda3/etc/profile.d/conda.sh
conda activate SE3nv
cd /(root)/mydesigns
python3 ../RFdiffusion/scripts/run_inference.py \
scaffoldguided.target_path=./target/target.pdb \
inference.output_prefix=backbones_OUT/ \
scaffoldguided.scaffoldguided=True \
$id scaffoldguided.target_pdb=True \
scaffoldguided.target_ss=./slightly_truncation_adj_secstruct/slightly_truncation_ss.pt \
scaffoldguided.target_adj=./slightly_truncation_adj_secstruct/slightly_truncation_adj.pt \
scaffoldguided.scaffold_dir=../RFdiffusion/examples/ppi_scaffolds/ \
scaffoldguided.mask_loops=False \
inference.num_designs=10000 \
denoiser.noise_scale_ca=0 \
denoiser.noise_scale_frame=0
Untar 1000 scaffold templates
Don't forget to untar the provided 1000 scaffold templates (RFdiffusion/examples/ppi_scaffolds_subset.tar.gz). In ppi_scaffolds_subset.tar.gz, there is ppi_scaffolds folder and we will use it in our backbone designing script.
Now, our directories tree becomes:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
Add ppi_scaffolds argument in backbone name
In RFdiffusion/scripts/run_inference.py, I modify some codes a little bit, so that, the backbone pdb file name will contain the `ppi_scaffolds` argument.

inference.num_designs=10000 binder backbones for ppi.hotspot_res=[A28-A25-A29-A26-A63] argument is A28-A25-A29-A26-A63_0.pdb.
3️⃣ sequence design and binder assessment script mpnn_af.slm
Perform a filtering step
At this moment, we could perform a filtering step after we get all potential binder backbones. For example, I only want backbones (orange cross) in one side (green circle). We could remove other useless backbones, which can help a lot in reducing the computation stress.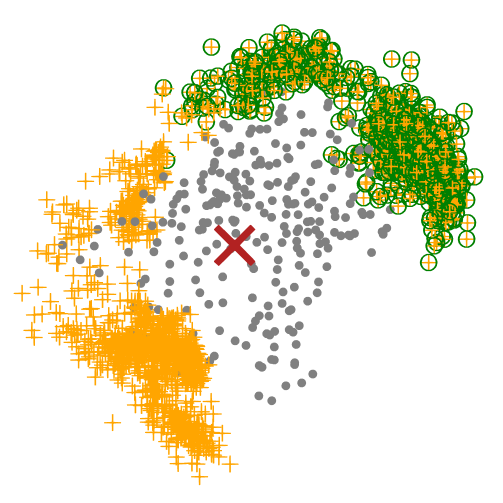
Let's say that we finally select 1000 favourite backbones, and we go to design binders based on these 1000 backbones. Normally, I will split favourite backbones into multiple folders, so I could parallelly design binders in each folder simultaneously. For example, in order to speed up the binder design process, I split the 1000 favourite backbones into 5 folders. After that, I could parallely execute #SBATCH -a 1-5 jobs simultaneously.
# sele_list contains names of 1000 selected backbones,
# such as A28-A25-A29-A26-A63_0.pdb
folder_size = 100 # how many pdb files you want at most to be grouped in one folder
chunks = [sele_list[x:x+folder_size] for x in range(0, len(sele_list), folder_size)]
# len(chunks)=5
for i in range(len(chunks)):
chunk = chunks[i]
newpath = "../mydesigns/select_1000_mpnn_af/folder" + str(i)
if not os.path.exists(newpath):
os.makedirs(newpath)
for j in chunk:
src = "../mydesigns/backbones_OUT/" + j
dst = "../mydesigns/select_1000_mpnn_af/folder" + str(i) + "/" + j
shutil.copyfile(src, dst)
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
#SBATCH -J array-job
#SBATCH -a 1-5
#SBATCH -o mpnn_af_1000.%A.%a.log
id_list="./folderID.txt"
id=`head -n $SLURM_ARRAY_TASK_ID $id_list | tail -n 1`
cd /(root)/mydesigns/target_1000_mpnn_af
cd $id
source ~/miniconda3/etc/profile.d/conda.sh
conda activate proteinmpnn_binder_design
silentfrompdbs *.pdb > silent.silent
python ../../../BinderDesign/dl_binder_design/mpnn_fr/dl_interface_design.py -silent silent.silent -outsilent seq.silent
conda deactivate
conda activate af2_binder_design
python ../../../BinderDesign/dl_binder_design/af2_initial_guess/predict.py -silent seq.silent -outsilent af2.silent -scorefilename score.sc
Now our directories tree is like:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
│ ├── select_1000_mpnn_af
│ │ ├── folder0
│ │ │ └── A28-A25-A29-A26-A63_0.pdb
│ │ │ └── ...
│ │ │ └── af2.silent
│ │ │ └── check.point
│ │ │ └── score.sc
│ │ │ └── seq.silent
│ │ │ └── seq.silent.idx
│ │ │ └── silent.silent
│ │ │ └── silent.silent.idx
│ │ ├── folder1
│ │ ├── ...
│ │ ├── folderID.txt
│ │ ├── mpnn_af.slm
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
4️⃣ Get designed binders
In each mydesigns/select_1000_mpnn_af/folder, af2.silent has our designed binders. And, we will extract their .pdb files from the .silent file by (for example)
#!/bin/bash
#SBATCH -q gpu-huge
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=8
#SBATCH -p GPU-name
#SBATCH --gres=gpu:1
#SBATCH --mem=50G
cd /(root)/mydesigns/select_1000_mpnn_af/folder0
source ~/miniconda3/etc/profile.d/conda.sh
conda activate proteinmpnn_binder_design
silentextract af2.silent
Our final directories tree is like:
├── dl_binder_design
│ ├── af2_initial_guess
│ │ ├── predict.py
│ ├── mpnn_fr
│ │ ├── dl_interface_design.py
├── mydesigns
│ ├── get_adj_secstruct.slm
│ ├── paraID.txt
│ ├── bb.slm
│ ├── target
│ │ └── target.pdb
│ ├── target_adj_secstruct
│ │ └── target_adj.pt
│ │ └── target_ss.pt
│ ├── backbones_OUT
│ │ ├── traj
│ │ ├── A28-A25-A29-A26-A63_0.pdb
│ │ ├── A28-A25-A29-A26-A63_0.trb
│ │ ├── ...
│ ├── select_1000_mpnn_af
│ │ ├── folder0
│ │ │ └── A28-A25-A29-A26-A63_0.pdb
│ │ │ └── A28-A25-A29-A26-A63_0_dldesign_0_cycle1_af2pred.pdb
│ │ │ └── ...
│ │ │ └── af2.silent
│ │ │ └── check.point
│ │ │ └── score.sc
│ │ │ └── seq.silent
│ │ │ └── seq.silent.idx
│ │ │ └── silent.silent
│ │ │ └── silent.silent.idx
│ │ ├── folder1
│ │ ├── ...
│ │ ├── folderID.txt
│ │ ├── mpnn_af.slm
├── RFdiffusion
│ ├── rfdiffusion
│ ├── scripts
│ │ └── run_inference.py
│ ├── helper_scripts
│ │ └── make_secstruc_adj.py
Each file with _cycle1_af2pred.pdb in name is our expected designed binders.