Random Notes
large-language model (LLM)
Recently, I read some papers, and I found one commonality that they treat protein data as text data. The natural language processing (NLP) methods seem highly transferable to the computational biology. For example, the Tansformer model is specially effective for language understanding, such as the language translation. ESM3 smartly used the mechanisms of Transformer in its own model and specially for the protein task.
A consensus is developing that underlying these sequences is a fundamental language of protein biology that can be understood using language models (6–11)
I would like to keep sharing what I am learning.
- Hayes, Tomas, et al. "Simulating 500 million years of evolution with a language model." bioRxiv (2024): 2024-07.
Simulating 500 million years of evolution with a language model
article
In Introduction section, authors state why they started this work. Firstly, architectures and representations within language models, especially transformer-based ones, map very well onto the world of protein. Secondly, "Great strength produces miracles". Increasing scale means increasing accuracy.
A number of language models of protein sequences have now been developed and evaluated (5–10, 12–17). It has been found that the representations that emerge within language models reflect the biological structure and function of proteins (6–8, 18), and are learned without any supervision on those properties (19, 20), improving with scale (5, 21). In the field of artificial intelligence, scaling laws have been found that predict the growth in capabilities with increasing scale, describing a frontier in compute, parameters, and data.
So, they give the ESM3 model, a multimodal generative language model (sequence, strcuture, function) . It was trained on 2.78 billion proteins, 771 billion unique tokens, and has 98 billion parameters.
After the ESM3 is trained (training process is supervised), they set up an input sequence, and asked the ESM3 model to generate a group of protein who are much likely to achieve in lab experiments.
We set out to create a functional green fluorescent protein (GFP) with low sequence similarity to existing ones. We chose the functionality of fluorescence because it is difficult to achieve, easy to measure, and one of the most beautiful mechanisms in nature.
They then performed a first experiment with 88 designs.
Among the generations that we synthesized, we found a bright fluorescent protein at far distance (58% identity) from known fluorescent proteins. Similarly distant natural fluorescent proteins are separated by over five hundred million years of evolution
The SEM3, a multi-modal transformer-based generative model, makes it true to find or design new proteins based on certain conditions.
We have found that language models can reach a design space of proteins that is distant from the space explored by natural evolution, and generate functional proteins that would take evolution hundreds of millions of years to discover. Protein language models do not explicitly work within the physical constraints of evolution, but instead can implicitly construct a model of the multitude of potential paths evolution could have followed
notes
What excits me a lot in this article? (I am thinking these points might be used in my tasks.)
- the way of integrating sequence, structure and function together.
- the application of transformer model.
I spend some time on the Transformer model. In the following example, it is a translation from lets go to vamos.
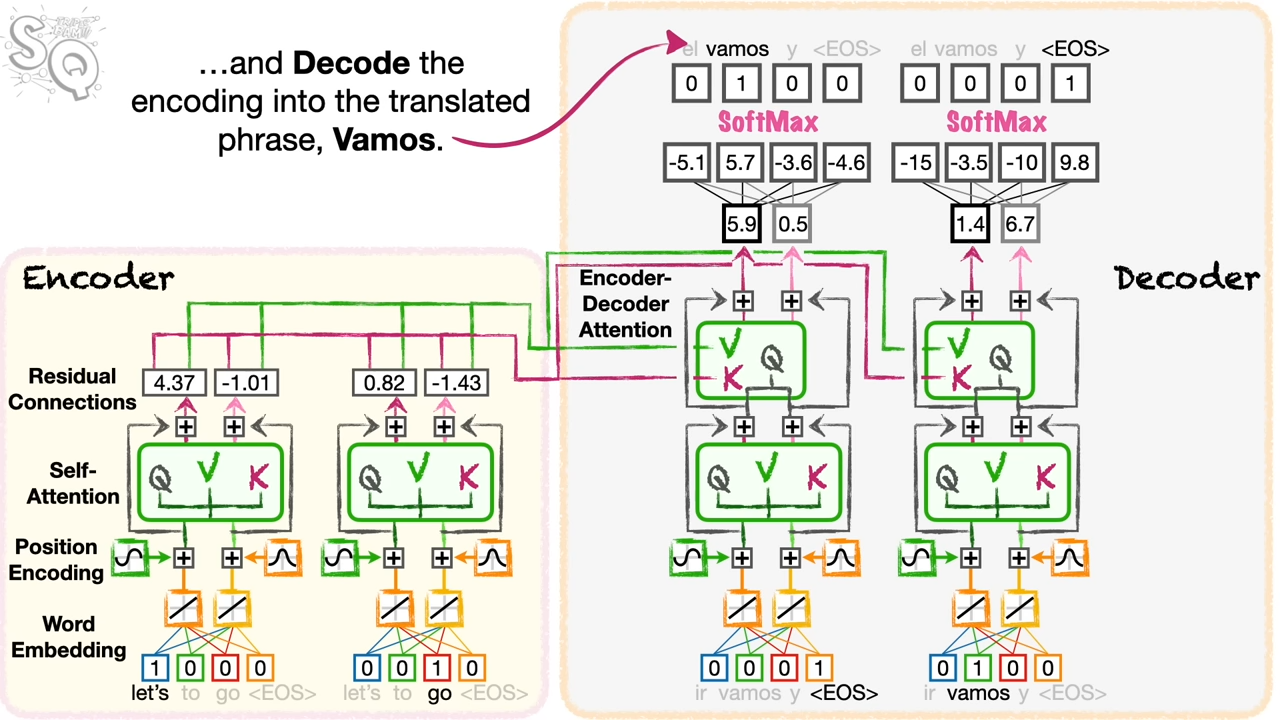
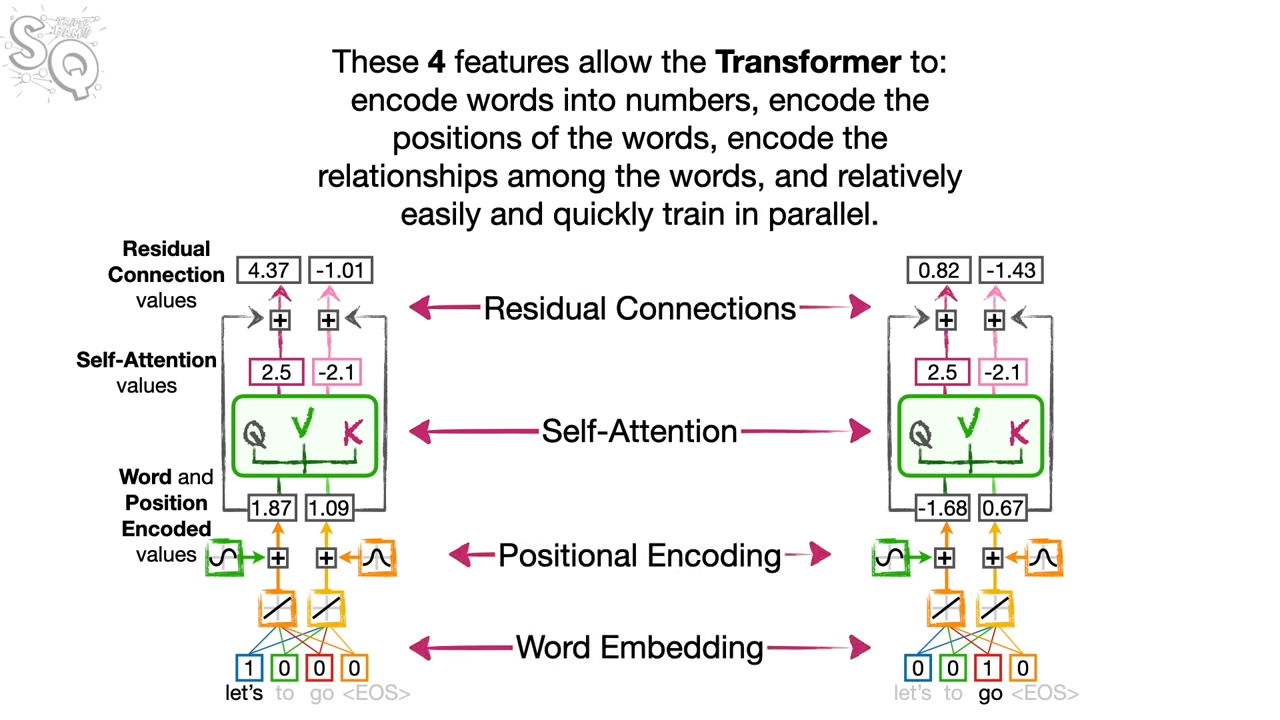
domain definition
If I have protein dataset (fasta and pdb files), and I can perform cluster analysis using similarity calculation, then, how to define or decide to say this set of (parts of) sequences is much worthy for us to do wet-lab experiments, in order to discover new domains.
- discover uncharacterized proteins with targe domains
- discover uncharacterized proteins purely using cluster and alignment analysis
SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures
1. Discover uncharacterized proteins with targe domains
Example
Structure-guided discovery of ancestral CRISPR-Cas13 ribonucleases
replication
tools:
- ColdFold ( https://github.com/sokrypton/ColabFold ) for structure prediction.
- DaliLite.v5 manual ( http://ekhidna2.biocenter.helsinki.fi/dali/README.v5.html ) for systematic search (kind of structure-based search).
- HMMsearch ( http://hmmer.org/documentation.html ) for sequence similarity search.
- CRISPR Recognition Tool (CRT) ( https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-8-209 ) http://www.room220.com/crt/
progress:
- Cas13a, Cas13b, and Cas13d whose structures are PDBID: 5XWP, 6DTD, 6E9F
- For Cas13c, which lacks an experimental structure, a ColabFold (43) model of A0A9X2MGT7 was generated using three recycles without amber relaxation. A0A9X2MGT7 fasta is https://www.uniprot.org/uniprotkb/A0A9X2MGT7/entry
ColabFold Usage to generate A0A9X2MGT7 structure
# https://github.com/sokrypton/ColabFold
colabfold_batch A0A9X2MGT7.fasta out_dir --num-recycle 3
# To activate this environment, use
#
# $ conda activate /storage/shenhuaizhongLab/lihuilin/ColabFold/localcolabfold/colabfold-conda
#
# To deactivate an active environment, use
#
# $ conda deactivate
Installation of ColabFold finished.
Add /storage/shenhuaizhongLab/lihuilin/ColabFold/localcolabfold/colabfold-conda/bin to your PATH environment variable to run 'colabfold_batch'.
i.e. for Bash:
export PATH="/storage/shenhuaizhongLab/lihuilin/ColabFold/localcolabfold/colabfold-conda/bin:$PATH"
For more details, please run 'colabfold_batch --help'.
(/storage/shenhuaizhongLab/lihuilin/ColabFold/localcolabfold/colabfold-conda) [lihuilin@gvnq04 localcolabfold]$ colabfold_batch A0A9X2MGT7.fasta out_dir --num-recycle 3
2024-08-07 11:04:22,702 Running colabfold 1.5.5 (1ccca5a53d20c909f3ccf8a4b81df804e6717cb1)
WARNING: You are welcome to use the default MSA server, however keep in mind that it's a
limited shared resource only capable of processing a few thousand MSAs per day. Please
submit jobs only from a single IP address. We reserve the right to limit access to the
server case-by-case when usage exceeds fair use. If you require more MSAs: You can
precompute all MSAs with `colabfold_search` or host your own API and pass it to `--host-url`
2024-08-07 11:04:23,783 Running on GPU
2024-08-07 11:04:24,437 Found 5 citations for tools or databases
2024-08-07 11:04:24,437 Query 1/1: tr_A0A9X2MGT7_A0A9X2MGT7_9FIRM_Type_VI-C_CRISPR-associated_RNA-guided_ribonuclease_Cas13c_OS_Anaerosalibacter_massiliensis_OX_1347392_GN_cas13c_PE_4_SV_1 (length 1111)
COMPLETE: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 150/150 [elapsed: 00:01 remaining: 00:00]
2024-08-07 11:04:27,519 Setting max_seq=64, max_extra_seq=1
2024-08-07 11:05:56,561 alphafold2_ptm_model_1_seed_000 recycle=0 pLDDT=55.2 pTM=0.45
2024-08-07 11:06:50,394 alphafold2_ptm_model_1_seed_000 recycle=1 pLDDT=68.6 pTM=0.646 tol=11.5
2024-08-07 11:07:44,227 alphafold2_ptm_model_1_seed_000 recycle=2 pLDDT=73.1 pTM=0.686 tol=2.22
2024-08-07 11:08:38,082 alphafold2_ptm_model_1_seed_000 recycle=3 pLDDT=73 pTM=0.685 tol=0.964
2024-08-07 11:08:38,083 alphafold2_ptm_model_1_seed_000 took 231.7s (3 recycles)
2024-08-07 11:09:32,989 alphafold2_ptm_model_2_seed_000 recycle=0 pLDDT=61.8 pTM=0.491
2024-08-07 11:10:26,871 alphafold2_ptm_model_2_seed_000 recycle=1 pLDDT=71.2 pTM=0.655 tol=7.3
2024-08-07 11:11:20,753 alphafold2_ptm_model_2_seed_000 recycle=2 pLDDT=73.8 pTM=0.698 tol=2.15
2024-08-07 11:12:14,653 alphafold2_ptm_model_2_seed_000 recycle=3 pLDDT=73.9 pTM=0.695 tol=0.867
2024-08-07 11:12:14,655 alphafold2_ptm_model_2_seed_000 took 215.5s (3 recycles)
2024-08-07 11:13:09,562 alphafold2_ptm_model_3_seed_000 recycle=0 pLDDT=64.3 pTM=0.554
2024-08-07 11:14:03,448 alphafold2_ptm_model_3_seed_000 recycle=1 pLDDT=72.8 pTM=0.688 tol=7.73
2024-08-07 11:14:57,322 alphafold2_ptm_model_3_seed_000 recycle=2 pLDDT=75.1 pTM=0.712 tol=1.73
2024-08-07 11:15:51,181 alphafold2_ptm_model_3_seed_000 recycle=3 pLDDT=76.1 pTM=0.716 tol=0.902
2024-08-07 11:15:51,182 alphafold2_ptm_model_3_seed_000 took 215.5s (3 recycles)
2024-08-07 11:16:46,085 alphafold2_ptm_model_4_seed_000 recycle=0 pLDDT=62.3 pTM=0.528
2024-08-07 11:17:39,954 alphafold2_ptm_model_4_seed_000 recycle=1 pLDDT=73.9 pTM=0.709 tol=6.2
2024-08-07 11:18:33,819 alphafold2_ptm_model_4_seed_000 recycle=2 pLDDT=75.2 pTM=0.718 tol=1.76
2024-08-07 11:19:27,693 alphafold2_ptm_model_4_seed_000 recycle=3 pLDDT=75.3 pTM=0.708 tol=1.54
2024-08-07 11:19:27,695 alphafold2_ptm_model_4_seed_000 took 215.5s (3 recycles)
2024-08-07 11:20:22,592 alphafold2_ptm_model_5_seed_000 recycle=0 pLDDT=64.3 pTM=0.591
2024-08-07 11:21:16,475 alphafold2_ptm_model_5_seed_000 recycle=1 pLDDT=73.8 pTM=0.716 tol=6.65
2024-08-07 11:22:10,378 alphafold2_ptm_model_5_seed_000 recycle=2 pLDDT=74.9 pTM=0.731 tol=1.27
2024-08-07 11:23:04,285 alphafold2_ptm_model_5_seed_000 recycle=3 pLDDT=75.8 pTM=0.738 tol=1.16
2024-08-07 11:23:04,287 alphafold2_ptm_model_5_seed_000 took 215.6s (3 recycles)
2024-08-07 11:23:05,290 reranking models by 'plddt' metric
2024-08-07 11:23:05,309 rank_001_alphafold2_ptm_model_3_seed_000 pLDDT=76.1 pTM=0.716
2024-08-07 11:23:05,310 rank_002_alphafold2_ptm_model_5_seed_000 pLDDT=75.8 pTM=0.738
2024-08-07 11:23:05,311 rank_003_alphafold2_ptm_model_4_seed_000 pLDDT=75.3 pTM=0.708
2024-08-07 11:23:05,312 rank_004_alphafold2_ptm_model_2_seed_000 pLDDT=73.9 pTM=0.695
2024-08-07 11:23:05,313 rank_005_alphafold2_ptm_model_1_seed_000 pLDDT=73 pTM=0.685
2024-08-07 11:23:08,499 Done
- isolate HEPN core regions using ChimeraX:
(1) 5XWP: Cas13a, HEPN domain sequences (3 fragments) extraction using ChimeraX
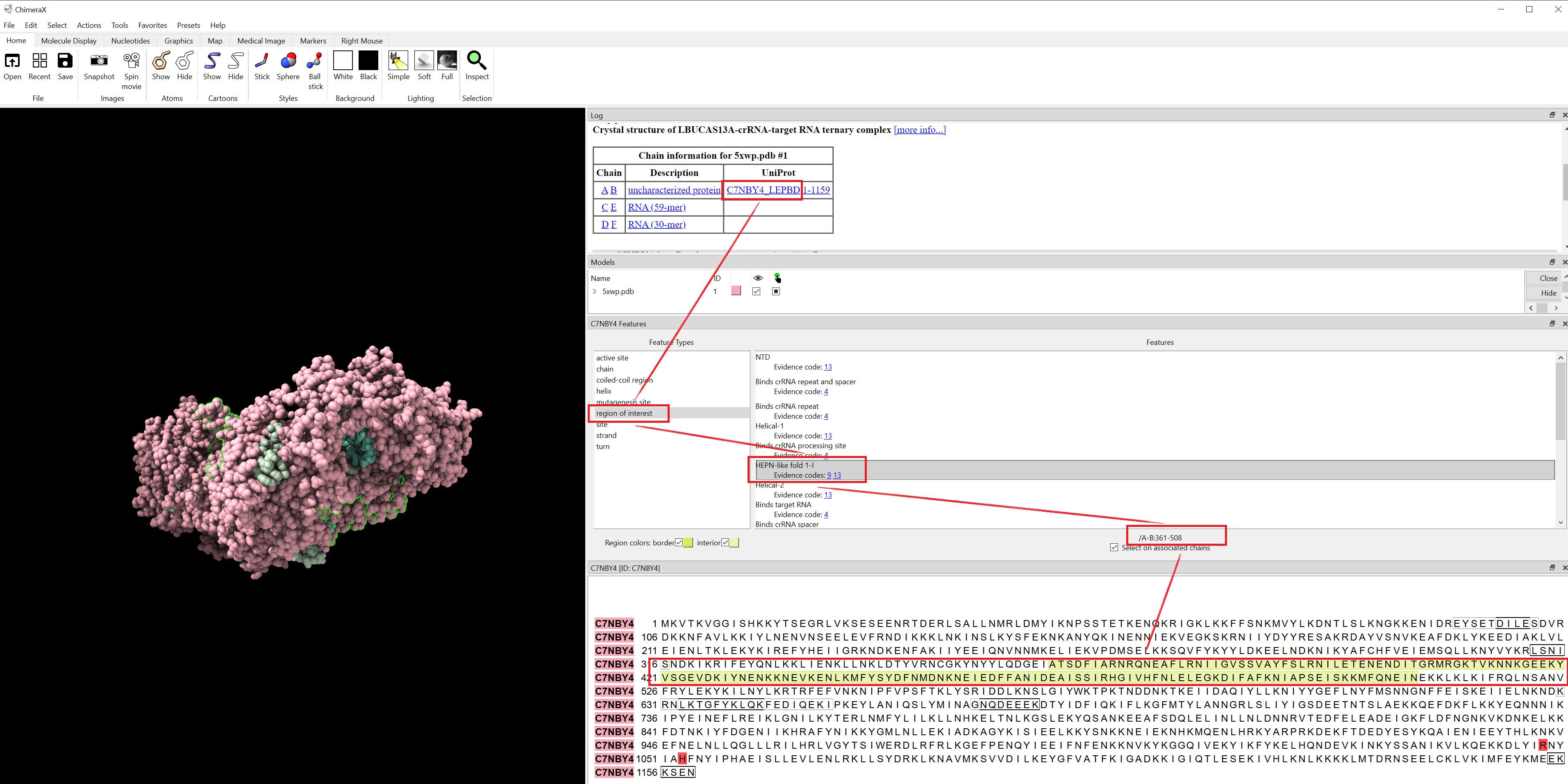
# cas13a-5xwp-HEPN-like-fold-1-I = 5xD1.pdb = sequence_ids = list(range(361,509))
# cas13a-5xwp-HEPN-like-fold-1-II = 5xD2.pdb = sequence_ids = list(range(752,814))
# cas13a-5xwp-HEPN-like-fold-2 = 5xD3.pdb = sequence_ids = list(range(947,1153))
(2) 6DTD:
(3) 6E9F:
 (4) A0A9X2MGT7:
(4) A0A9X2MGT7:
- All pdb to internal data format (xxxxX.dat) used by Dali/import.pl
$ tar -zxvf DaliLite.v5.tar.gz, $ cd DaliLite.v5/bin, $ make clean, $ make, replace $ DaliLite.v5/bin/dsspcmbi with $ aliLite.v5/bin/dsspcmbi/dsspcmbi
- systematic search by Dali/dali.pl
- filter by Z-score
- filter by signature Rx4H motif of HEPN domain. (sequence level analysis)
- results of dali as new queries,sequence-based search via PSI-BLAST and HMMsearch, new candidates, to collect highly matchable candidates-2
original text
Following this sequence-based enrichment step, contigs encoding the Cas13 candidates were downloaded, and annotated for CRISPR arrays using the CRT tool (v1.2) (48). This analysis led to the identification of the Cas13an systems characterized in this study.
-
Customized places:
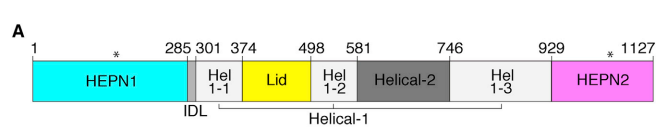
1.1 target example (HEPN domain in Cas13a, Cas13b, Cas13c, Cas13d) 1.2 target features (Rx4H motif, R****H) -
Firstly structure-level search; Secondlly sequence-level search.
-
The focus is on significant discoveries rather than methods.
############################################## stop here #############################################################
module load blast/2.11.0+
cd domainPDB
../bin/import.pl
--pdbfile ../toy_PDB/pdb1ppt.ent
--pdbid 1ppt
--dat ./
> import.stdout 2> import.stderr
../bin/dali.pl
--cd1 2nrmA # --cd1 <xxxxX> query structure identifier
--db pdb.list #--db <filename> list of target structure identifiers
--TITLE systematic
--dat1 ../DAT # path to directory containing query data [default: ./DAT/]
--dat2 ../DAT # path to directory containing target data [default: ./DAT/]
> systematic.stdout
2> systematic.stderr
- http://ekhidna2.biocenter.helsinki.fi/dali/README.v5.html
###############################################################
- automated structureal-search pipeline
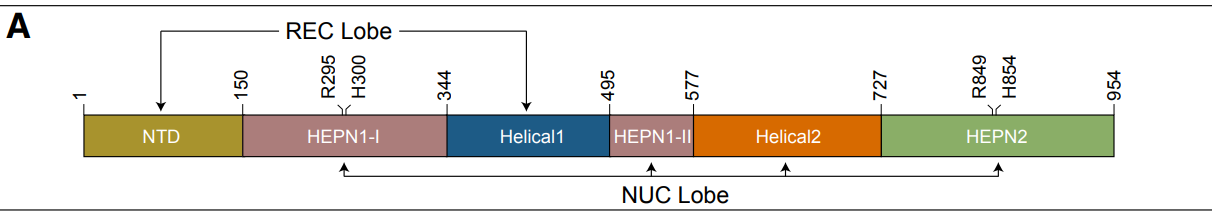
- identify an ancestral clade of Cas13 (Cas13an).
- trace Cas13 origins to defense-associated ribonucleases.
- Cas13an is 1/3 size of other Cas13s
- Cas13an mediates: 1) robust programmable RNA depletion and 2) defense against diverse bacteriophages
- pipeline:
- leveraged a Fold seek-clustered AlphaFold database (20), whose reduced search space makes slow-but-sensitive DALI searches feasible. Preprocessed AlphaFold Database for use with DaliLite running locally: link
- Using representative HEPN dimers within known Cas13 proteins (21–23) as the search query, we found twelve previously uncharacterized protein clusters in the AlphaFold database bearing an intramolecular HEPN dimer (fig. S1 and tables S1 and S2).
- Further sequence-based homology searches and genomic analyses revealed that two of the newly identified clusters occur next to CRISPR arrays, representing a new Cas13 subtype (Cas13an) (Fig. 1A and fig. S2)
- Notably, neither Foldseek nor hidden Markov model searches were able to detect significant homology between previously known Cas13s and Cas13an (fig. S3).
- This highlights the considerable divergence of Cas13an compared to known Cas13 proteins, and underscores the importance of sensitive search strategies.
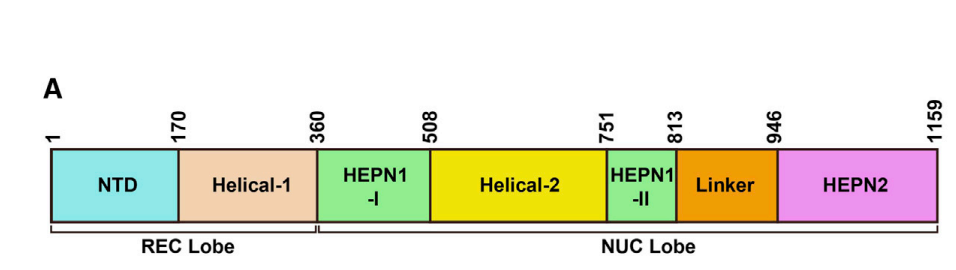
Structural-search and novel CRISPR system identification pipeline
- By manually examining the structures of previously characterized Cas13 subtypes, researchers curated the conserved intramolecular HEPN dimer core. (as the conserved domain)
- get 4 examples (Cas13a, Cas13b, and Cas13d, and Cas13c)
- The HEPN core regions were isolated from the Cas13 protein structures using ChimeraX (44).
- The HEPN core regions are used as queries in DALI.
To search against the entirety of the AlphaFold database (~200 million structures) (14), DALI’s slow performance presented a major bottleneck. To circumvent this, the search was performed against the ~2.3 million cluster representatives generated by clustering the AlphaFold database with Foldseek
- ~2.3 million cluster representatives were subdivided into batches that each contained 1000 structures, resulting in a total of 2304 batches.
- After completing DALI-searches against the AlphaFold database, only hits with the signature Rx4H motif of HEPN domains and a DALI Z-score greater than 6 were retained.
- Finally, only hits that also bear an intramolecular HEPN dimer were considered as Cas13 candidates.
- Sequences of Cas13 candidates were retrieved and used as input for PSI-BLAST (45) and HMMsearch (46) against the NCBI NR database (47) using default settings.
- contigs encoding the Cas13 candidates were downloaded, and annotated for CRISPR arrays using the CRT tool (v1.2) (48). This analysis led to the identification of the Cas13an systems characterized in this study.
- 5XWP:
2. discover uncharacterized proteins purely using cluster and alignment analysis
Holm, Liisa, et al. "DALI shines a light on remote homologs: One hundred discoveries." Protein Science 32.1 (2023): e4519.
-
Pfam 35.0, which contains a total of 19,632 families. Among these, 7770 families are grouped into 655 clans, corresponding to superfamilies in other classifications (Mistry et al., 2020).
-
Many Pfam families represent domains, covering only part of a protein.
-
For each Pfam family, we selected a representative member of median length for that family.
-
Pfam 35.0 has 19,632 families.
- select 59% who are in AlphaFold Database version 1
- cut-off pLDDT<70, resulting in cropped models
- (BECAUSE: at not all AlphaFold models are compact and globular)
- filter0: remove many ill-folded models from the analysis as confirmed by visual inspection.
- filter2: the contact order (CO) had to meet the range of native proteins (CO > 0.05)
- the model had to exhibit at least 60 confidently modeled residues represented by at least four secondary structure elements, that is, either alpha helices or beta strands.- (BECAUSE: Pfam provides public tables that link (i) PDB structures to protein families, and (ii) distantly related families to clans.)
- remove families that have a member with a PDB structure.
- remove families that having a clan member with a PDB structure.
----> 1412 families without known structures.
- (BECAUSE: Pfam provides public tables that link (i) PDB structures to protein families, and (ii) distantly related families to clans.)
- select 59% who are in AlphaFold Database version 1
enzyme
Enhanced crystalline cellulose degradation by a novel metagenome-derived cellulase enzyme
Celcm05-2: The camel rumen metagenome-assembled sequences and the predicted protein-coding genes have been deposited to the Integrated Microbial Genomes (IMG) database under the IMG dataset ID: 3300003523. link
- a broader pH range

slides












Cell-free chemoenzymatic starch synthesis from carbon dioxide
Modules:
- C1 module (for formaldehyde production),
- C3 module (for D-glyceraldehyde 3-phosphate production),
- C6 module (for D-glucose-6-phospate production),
- Cn module (for starch synthesis).
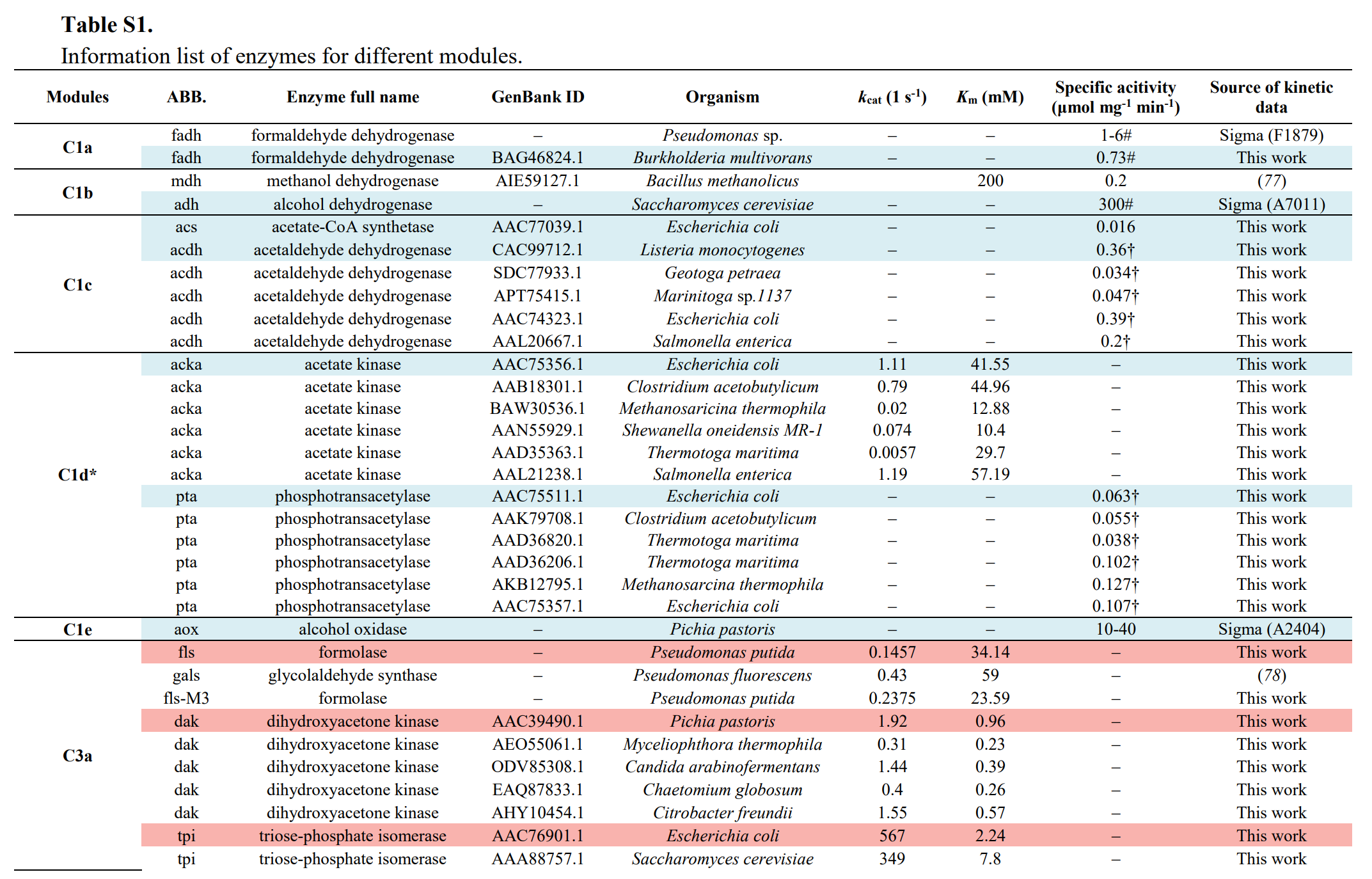
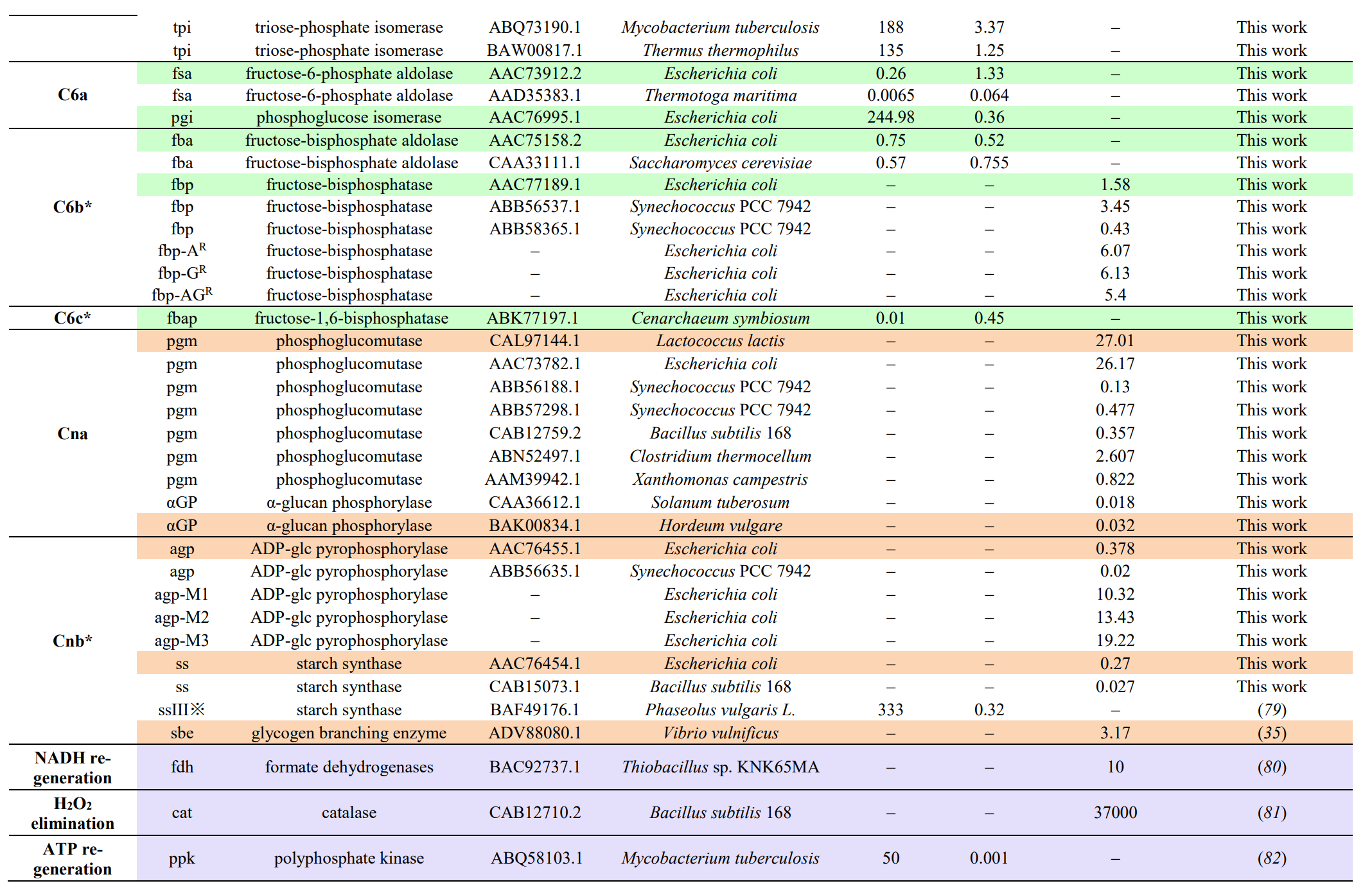

Enzymes selectioin:
- balance between conversion rate and enzyme yield.
- inhibition between enzymes.
- variants




FLSHclust
Altae-Tran, Han, et al. "Uncovering the functional diversity of rare CRISPR-Cas systems with deep terascale clustering." Science 382.6673 (2023): eadi1910.
Data
sequence data in fasta format
Algorithm overview
- Input the original data.
- Initial analysis to find most likely similar pairs.
- e.g. we have 1000 sequence, there should have 499,500 pairs for comparisons. The work of this initial analysis is to narrow these pairs down to 6041 pairs.
- reproduced well.
- more details about this step are as follows.
- has been verifyed that the final 19 pairs come from these 6041 pairs.
- The next analysis is based on these this reduced pairs.
- These reduced pairs are randomly assigned to 32 partitions in PySpark.
- Similarity calculation is performed in these partitions concurrently.
- similarity calculate:
# 1 parasail.sw_trace_scan_16(x, y, 11, 1, parasail.blosum62) # 2 edlib.align(x, y, mode='HW', task='locations', k=max_dist + 1) # edit distance- filter again based on one threshold
min_seq_id. - potential pairs are narrowed down again from 6041 to 21.
- yes, the final 19 pairs all come from these 21 pairs.
1000 seq (~500K pairs) ---step1---> 6041 pairs ---step2---> 21 pairs
- In
step1, LSH algorithm is the core role. It reduces the number of comparisons to a few thousands. - In
step2, the common similarity calculation method is performed.
Why the cluster work is already done at this moment? If we check these 21 pairs and the final 19 pairs carefully, wa can see the 21 pairs already are divided into 19 groups. And, the final 19 pairs come from each cluster, and pick the most similar pair if there are more than one pairs in each cluster. For example, if we talk about the similar sequence to Seq_173, we find two: Seq_715 and Seq_896, however, in the final output, we only pick the Seq_715 which is the most similar one.
| 19 pairs | 21 pairs |
|---|---|
| (173, 715) | (173,715), (173,896) |
| (889,582) | (889,582), (889,906) |
- If we continue their work, we can see their following work is to point out the only pairs who are most close to each other in each cluster.
- That is the reason of why we only see two similar sequence in each cluster in the final output file.
LSH work reproduce
./MYANAWORK/
└── all_data.fa
└── smallflsh
└── __init__.py
└── functions.py
└── read_to_parquet(fastapath, parquestpath)
├── myflsh.py
├── sample1k.fa
├── smallflsh
│ ├── functions.py
│ | ├── read_to_parquet(fastapath, parquestpath)
│ ├── get_kmers.py
│ ├── __init__.py
├── target.fa
outline
-
read into [
ID,aa_seq] -
till now, resulting in
a set of protein IDs sorted by its possibility of being closer to target protein.
Application in 11M sequences
outofmemory when performing PySpark calculation. --> incorrectly use Spark --> change to use dask-cuDF, works
ongoing
dataset
ftp.ensembl.org
https://ftp.ensembl.org/pub/current_fasta/
size(no duplicates): 11,551,135 (11 million)
# get .gz files
wget -r -np -e robots=off -A 'pep.all.fa.gz' https://ftp.ensembl.org/pub/current_fasta/
import os, gzip, shutil
import glob
import numpy as np
import pandas as pd
from Bio import SeqIO
# unzip all .gz files
start_dir = "./ftp.ensembl.org/pub/current_fasta/"
gz_count = []
destination_dir = "./fa_save/"
for root, dirs, files in os.walk(start_dir):
for gz_name in files:
gz_file = os.path.join(root, gz_name)
if "/pep/" in gz_file:
# print(gz_file)
gz_count.append(gz_file)
out_file = destination_dir + gz_name.split(".gz")[0]
with gzip.open(gz_file,"rb") as f_in, open(out_file,"wb") as f_out:
shutil.copyfileobj(f_in, f_out)
# read
with open("./fa_save/all_data.fa") as fasta_file:
identifiers = []
seq = []
for seq_record in SeqIO.parse(fasta_file, 'fasta'):
identifiers.append(seq_record.id)
seq.append("".join(seq_record.seq))
df_pro = pd.DataFrame()
df_pro["aa_seq"] = seq
df_pro["p_id"] = identifiers
# clear duplicates
df_pro_drop_dup = df_pro.drop_duplicates(subset='aa_seq')
Encoding methods
Encoding the raw data is the first thing we need to think of when we want to use ML for analyzing.
Encoding is the process of transforming raw data (text, images, audio) into a structured, numerical format that computers can readily process.
If we take protein sequence data as the example,
This is a way I learned from one paper.
Altae-Tran, Han, et al. "Uncovering the functional diversity of rare CRISPR-Cas systems with deep terascale clustering." Science 382.6673 (2023): eadi1910.
random biology knowledge
Dali
some assessment criteria
sequence comparison
query coverage sequence identify
some vocab
consensus sequence
sequence logos
sequence logos方法利用每个位置的高度来表示其保守程度,这样特征高度就反映了相对的变化频率,比如说一个consensus sequence位置上可能是C或者T,sequence logos上C的高度代表了其通常比T出现的频率多5倍。
vocabulary and expression
Glossary
| term | knowledge |
|---|---|
| alleles | 等位基因, a gene can exit in different forms called alleles. e.g. the pea can have either yellow or green seeds. One allele of the gene for seed color gives rise to yellow seeds, the other to green. Moreover, one allele can be dominant over the other, recessive, allele. |
| dominat, recessive | 显性, 隐性 |
| transmission genetics | 传播遗传学 |
| blending | 混合 |
| progeny | 后代 |
| phenotype | 表型, observable characteristic, Greek root: phemomenon, meaning appearance. exhibit the tall phenotype/appearance. refer to the whole set of observable characteristics of an organism |
| filial generation F1, F2 ... | 子代, filial comes from the Latin: filius=son, filia=daughter. |
| self-fertilize | 自花授粉 |
| diploid | 二倍体, the parents were diploid |
| homozygotes | 纯合子,homozygotes have two copies of the same allele. |
| heterozygotes | 杂合子, heterozygotes have one copy of each allele. |
| haploid | 单倍体, sex cells contain only one copy of the gene; that is, thay are haploid. |
| gametes | 配子, or sex cells |
| cell nucleus | 细胞核 |
Expression
- inheritance is particulate
- each parent contributes particles, or genetic units, to the offspring. We now call these particles genes.
- the allele for green seeds must have been preserved in the F1 generation.
Names
| this | is | and |
|---|---|---|
| Dropsopbila melanpgaster | fruit fly |
commands
stat_gpu # check available partition
srun -q gpu -p v100-af --mem=50G --gres=gpu:1 --pty /bin/bash # access one gpu
tar xvzf file.tar.gz # unzip
module load igv # access specific place
